El síndrome del X frágil es una enfermedad genética ligada al cromosoma X que constituye, después del síndrome de Down, la causa más frecuente de discapacidad intelectual en varones. Aproximadamente 1 de cada 4,000 hombres presenta esta condición. Se caracteriza por un fenotipo neurológico complejo que incluye manifestaciones dentro del espectro autista, impulsividad, comportamientos repetitivos y, en algunos casos, agresividad.
Manifestaciones clínicas
Esta alteración genética también puede afectar la función intelectual en mujeres, aunque con menor severidad y con una frecuencia aproximadamente 50% inferior a la observada en varones. Las mujeres portadoras heterocigotas suelen no presentar signos físicos evidentes, aunque pueden experimentar dificultades en el aprendizaje, trastornos de ansiedad, hipersensibilidad sensorial o incluso cierto grado de discapacidad intelectual. Además, es frecuente que desarrollen menopausia precoz como único signo clínico visible.
En los varones afectados, las manifestaciones físicas son más claras. Tras la pubertad, se observa macroorquidismo (aumento del tamaño de los testículos), además de orejas prominentes, mandíbula saliente, voz aguda y rasgos conductuales del espectro autista, junto con retraso mental. Algunos de estos individuos también presentan signos de un trastorno leve del tejido conectivo, como hipermovilidad articular y prolapso de la válvula mitral.
Las personas portadoras de la premutación del gen FMR1 —es decir, aquellas con entre 55 y 200 repeticiones del triplete CGG— están expuestas a riesgos específicos. Las mujeres con esta premutación tienen una mayor probabilidad de desarrollar insuficiencia ovárica prematura (conocida como FXPOI) y pueden presentar alteraciones cognitivas leves. Tanto hombres como mujeres portadores de la premutación tienen un riesgo incrementado de padecer trastornos del ánimo y de ansiedad, así como de desarrollar, a partir de la mediana edad, un síndrome caracterizado por temblores y ataxia, denominado síndrome de temblor/ataxia asociado al X frágil (FXTAS).
En muchos casos, este síndrome se asocia con alteraciones cerebrales visibles mediante estudios de imagen, especialmente en la sustancia blanca del cerebelo, incluso antes de que aparezcan los síntomas clínicos. Dado que la prevalencia de portadores de la premutación en la población general es relativamente alta (entre 1 de cada 130 y 1 de cada 600 individuos), se recomienda realizar pruebas genéticas dirigidas al análisis del locus FMR1 en personas mayores que presenten alteraciones neurológicas o conductuales compatibles con esta condición.
Exámenes diagnósticos
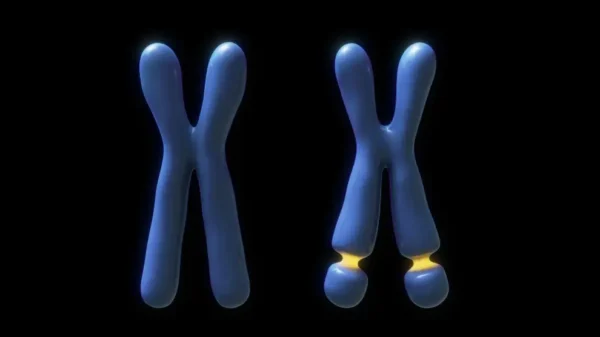
El síndrome del X frágil fue inicialmente identificado gracias a una alteración citogenética característica: una zona de fragilidad visible como una pequeña interrupción o estrechamiento en el extremo del brazo largo del cromosoma X. Esta anomalía estructural llamó la atención de los genetistas, ya que coincidía de manera consistente con individuos que presentaban discapacidad intelectual. Más adelante, los avances en biología molecular permitieron descubrir que esta fragilidad estaba relacionada con una expansión anormal de una secuencia repetitiva de tres nucleótidos —citosina-guanina-guanina (CGG)— ubicada cerca del gen denominado FMR1 (del inglés fragile X mental retardation 1), cuya función es esencial para el desarrollo y funcionamiento del sistema nervioso.
En condiciones normales, todas las personas poseen una cierta cantidad de repeticiones del triplete CGG en esa región del cromosoma X, típicamente en un rango que no genera consecuencias clínicas. Sin embargo, cuando el número de repeticiones supera las 52 unidades, se incrementa la inestabilidad genética durante la formación de gametos, es decir, durante la espermatogénesis en los varones o la ovogénesis en las mujeres. Esta inestabilidad favorece una expansión progresiva del número de repeticiones en las siguientes generaciones, un fenómeno conocido como anticipación genética. Este proceso explica por qué la enfermedad puede aparecer con mayor gravedad o a una edad más temprana en la descendencia.
Cuando una persona nace con una copia del gen FMR1 que contiene 200 o más repeticiones del triplete CGG, se produce una alteración funcional significativa del gen. En la mayoría de los varones, esta condición se manifiesta con algún grado de discapacidad intelectual, mientras que en las mujeres, debido a la presencia de un segundo cromosoma X funcional, las consecuencias pueden ser más variables. Aproximadamente un 60% de las mujeres con esta expansión completa presentan algún nivel de deterioro cognitivo.
El número de repeticiones influye no solo en la severidad del cuadro clínico, sino también en la probabilidad de que se produzcan expansiones adicionales durante la formación de los gametos. Así, las familias con antecedentes de síndrome del X frágil pueden observar una progresión del trastorno a lo largo de las generaciones, tanto en intensidad como en frecuencia, debido a esta dinámica de inestabilidad genética que caracteriza al locus del gen FMR1.
Prevención
El diagnóstico del síndrome del X frágil ha evolucionado significativamente con el avance de las técnicas moleculares. En el pasado, la principal herramienta diagnóstica era el análisis citogenético, que permitía observar directamente el sitio frágil característico en el extremo del brazo largo del cromosoma X. Este método, aunque útil en su momento, presentaba limitaciones importantes: requería cultivos celulares especializados, tenía baja sensibilidad en algunos casos y no permitía una cuantificación precisa del número de repeticiones del triplete CGG en el gen FMR1, el verdadero responsable de la enfermedad.
En la actualidad, la diagnosis molecular mediante análisis de ADN ha reemplazado por completo al enfoque citogenético en la práctica clínica y en el contexto del diagnóstico prenatal. Esta técnica permite determinar con exactitud el número de repeticiones CGG en el locus del gen FMR1, lo que resulta esencial para identificar tanto casos de mutación completa —asociados a manifestaciones clínicas graves— como estados de premutación, que aunque pueden ser asintomáticos en un inicio, implican riesgos reproductivos y neurológicos importantes. Además, esta prueba puede identificar casos de alelos intermedios, cuya estabilidad puede variar entre generaciones.
La precisión del análisis molecular lo convierte en una herramienta de primera línea en la evaluación de cualquier individuo, ya sea hombre o mujer, que presente discapacidad intelectual sin causa aparente. Su aplicación es especialmente valiosa en la infancia, ya que un diagnóstico temprano permite planificar intervenciones terapéuticas y educativas adaptadas a las necesidades del paciente, así como ofrecer asesoramiento genético a la familia.
En paralelo, se está considerando la implementación de programas de tamizaje neonatal basados en la detección de niveles anómalos de metilación en el promotor del gen FMR1. En los casos de mutación completa, la región que regula este gen suele encontrarse hipermetilada, lo que conlleva la inactivación de la transcripción y, por ende, la ausencia de la proteína FMRP, crucial para el desarrollo neuronal. La detección de esta hipermetilación desde el nacimiento abriría la posibilidad de iniciar intervenciones desde etapas muy tempranas del desarrollo, con el objetivo de mitigar las manifestaciones clínicas y mejorar el pronóstico funcional a largo plazo.
Tratamiento
Hasta la fecha, no existe un tratamiento capaz de corregir directamente la alteración genética que origina el síndrome del X frágil. Esta condición está provocada por una expansión anómala de repeticiones del triplete CGG en el gen FMR1, lo que conduce a la silenciamiento epigenético del gen y a la consiguiente ausencia o deficiencia de la proteína FMRP, fundamental para la regulación de la plasticidad sináptica y el desarrollo neurológico normal. Al tratarse de un trastorno monogénico con consecuencias epigenéticas complejas, las estrategias terapéuticas dirigidas a restaurar la función del gen o revertir su silenciamiento siguen siendo un reto científico.
Frente a esta limitación, la investigación biomédica ha centrado sus esfuerzos en comprender las consecuencias funcionales de la deficiencia de FMRP, particularmente en lo que respecta a la neurotransmisión y la regulación sináptica. Modelos murinos del síndrome del X frágil, en los cuales se ha inactivado el gen Fmr1, han permitido identificar alteraciones específicas en la actividad de ciertos receptores, como los del glutamato y el ácido gamma-aminobutírico (GABA), así como en vías de señalización intracelular relacionadas con la síntesis proteica neuronal. Estos hallazgos han orientado el desarrollo de tratamientos experimentales que buscan modular los desequilibrios neuroquímicos derivados de la falta de FMRP, en lugar de corregir directamente la mutación genética subyacente.
Actualmente, varios de estos enfoques farmacológicos se encuentran en fase de ensayo clínico. Se investigan, por ejemplo, compuestos que actúan sobre los receptores metabotrópicos del glutamato, con el objetivo de atenuar la sobreactivación de ciertas vías sinápticas que se ha observado en el modelo murino. Otros agentes están dirigidos a mejorar la inhibición neuronal mediada por GABA, que suele estar disminuida en personas afectadas.
Entre los fármacos que han mostrado cierto beneficio sintomático se encuentra el ácido valproico, un anticonvulsivo con propiedades estabilizadoras del estado de ánimo. Aunque no modifica la causa genética de la enfermedad, puede contribuir a reducir síntomas conductuales como la hiperactividad y el déficit de atención. Sin embargo, su uso debe considerarse solo después de haber probado tratamientos estándar, especialmente aquellos que ya cuentan con respaldo clínico en poblaciones con trastornos del neurodesarrollo.


Fuente y lecturas recomendadas:
- Hagerman RJ et al. Fragile X syndrome: lessons learned and what new treatment avenues are on the horizon. Annu Rev Pharmacol Toxicol. 2022;62:365. [PMID: 34499526]
- Protic DD et al. Fragile X syndrome: from molecular aspect to clinical treatment. Int J Mol Sci. 2022;23:1935. [PMID: 35216055]
- Salcedo-Arellano MJ et al. Fragile X syndrome and associated disorders: clinical aspects and pathology. Neurobiol Dis. 2020;136:104740. [PMID: 31927143]
- Spector E et al. Laboratory testing for fragile X, 2021 revision: a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021;23:799. [PMID: 33795824]
Aprende administración paso a paso

ADMINISTRACION DESDE CERO