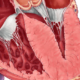
La miocardiopatía hipertrófica es una enfermedad genética que se hereda como un rasgo autosómico dominante, lo que implica que solo se necesita una copia alterada del gen para que se exprese la enfermedad en un individuo. Esta herencia autosómica dominante significa que un progenitor portador de la variante patogénica tiene un 50 por ciento de probabilidad de transmitir la condición a cada uno de sus hijos. Sin embargo, la penetrancia de este rasgo es variable, lo que indica que no todos los individuos portadores de la variante patogénica necesariamente desarrollarán la enfermedad. Esto puede depender de factores genéticos adicionales, ambientales, y posiblemente de la interacción con otros genes.
La miocardiopatía hipertrófica es causada por variantes patogénicas en uno de un gran número de genes, muchos de los cuales codifican para cadenas pesadas de miosina o para proteínas que regulan el manejo del calcio en el músculo cardíaco. Las cadenas pesadas de miosina son componentes fundamentales del sarcómero, la unidad contráctil del músculo cardíaco, y su alteración puede dar lugar a una contracción anormal. Las proteínas involucradas en la regulación del calcio, por su parte, son cruciales para el ciclo de contracción y relajación del corazón. Cualquier alteración en estos componentes puede resultar en el desarrollo de hipertrofia del ventrículo izquierdo, que es una característica central de la miocardiopatía hipertrófica.
El pronóstico de la enfermedad varía según la variante patogénica específica del gen implicado. Algunas variantes están asociadas con un riesgo mayor de complicaciones, como arritmias o muerte súbita, mientras que otras pueden estar asociadas con una progresión más benigna de la enfermedad. Esta variabilidad en el pronóstico resalta la importancia de un diagnóstico genético adecuado, que no solo puede confirmar la presencia de la miocardiopatía hipertrófica, sino también proporcionar información valiosa sobre el riesgo a largo plazo del paciente.
Los pacientes generalmente se presentan en la adultez temprana, aunque los síntomas pueden aparecer en diferentes momentos de la vida. En algunos casos, la enfermedad puede ser asintomática durante años, lo que complica su detección temprana. Es importante tener en cuenta que los síntomas pueden incluir disnea, palpitaciones y dolor en el pecho, que pueden ser confundidos con otras condiciones menos graves. Por lo tanto, el reconocimiento temprano de la miocardiopatía hipertrófica es crucial para su manejo y tratamiento adecuado, que puede incluir desde cambios en el estilo de vida hasta intervenciones médicas y quirúrgicas. La evaluación genética y el seguimiento continuo son fundamentales en la atención de los pacientes con esta enfermedad, dada su naturaleza hereditaria y los riesgos potenciales asociados.

Fuente y lecturas recomendadas:
- Olivotto I et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2020;396:759. [PMID: 32871100]
- Ommen SR. 2020 AHA/ACC guidelines for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: executive summary. Circulation. 2020;142:e533. [PMID: 33215938]
Aprende administración paso a paso

ADMINISTRACION DESDE CERO
Originally posted on 13 de octubre de 2024 @ 7:10 AM