
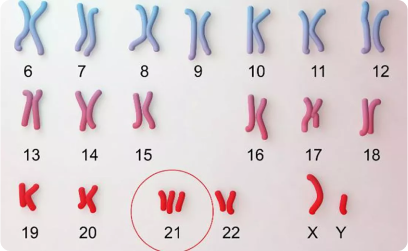
Aproximadamente el 0.5% de todas las concepciones humanas presentan trisomía del cromosoma 21, lo que significa que estas células poseen tres copias de dicho cromosoma en lugar de las dos habituales. Esta alteración genética, conocida como trisomía 21 o síndrome de Down, se produce principalmente como consecuencia de errores en la segregación cromosómica durante la meiosis, un fenómeno llamado no disyunción. Aunque esta frecuencia de concepción es relativamente alta, la incidencia de nacimientos vivos con síndrome de Down es notablemente menor, en torno a uno por cada setecientos nacimientos. Esta diferencia se debe a una elevada mortalidad fetal asociada a la trisomía 21, que impide el desarrollo completo de muchos embriones afectados.
El riesgo de concebir un hijo con síndrome de Down está fuertemente relacionado con la edad materna. En mujeres jóvenes, este riesgo se sitúa alrededor de uno por cada mil nacimientos; sin embargo, a medida que aumenta la edad de la madre, especialmente después de los treinta y cinco años, la probabilidad de anomalías cromosómicas se incrementa de forma pronunciada. Para una mujer de cuarenta y cinco años, por ejemplo, la probabilidad de tener un hijo con síndrome de Down puede alcanzar uno por cada cuarenta embarazos. Esta tendencia se debe a cambios acumulativos en los ovocitos femeninos, que permanecen en estado de detención meiótica durante décadas y, con el tiempo, son más susceptibles a errores en la distribución cromosómica. Dicho envejecimiento ovocitario conlleva una mayor probabilidad de no disyunción en la meiosis I o II. En contraste, la edad paterna avanzada no parece influir significativamente en la incidencia de trisomía 21.
Actualmente, existen métodos eficaces para detectar la presencia de trisomía 21 durante las primeras etapas del embarazo. Las pruebas de cribado en el primer y segundo trimestre incluyen la medición de biomarcadores en el suero materno, como la alfa-fetoproteína y otras proteínas específicas, conocidas colectivamente como pruebas de marcadores múltiples. Estas pruebas permiten estimar el riesgo de aneuploidías fetales. Además, la ecografía obstétrica puede identificar señales físicas indicativas del síndrome de Down, como el aumento del grosor nucal o el desarrollo insuficiente del hueso nasal, características que suelen estar presentes en fetos afectados.
Más recientemente, el análisis del ADN fetal libre en sangre materna ha revolucionado el diagnóstico prenatal no invasivo. Este enfoque, conocido como prueba prenatal no invasiva o detección de ADN fetal circulante, permite identificar con alta sensibilidad y especificidad anomalías cromosómicas como la trisomía 21, sin los riesgos asociados a procedimientos invasivos como la amniocentesis o la biopsia de vellosidades coriónicas.
Manifestaciones clínicas
El síndrome de Down suele ser identificado poco después del nacimiento debido a un conjunto distintivo de características clínicas observables. Entre los hallazgos físicos más comunes se encuentran rasgos craneofaciales característicos, como la braquicefalia (forma redondeada de la cabeza), la presencia de fisuras palpebrales oblicuas, el puente nasal plano, orejas de implantación baja y pliegues epicánticos. Además, los neonatos con esta condición suelen presentar hipotonía generalizada, es decir, una disminución del tono muscular que se manifiesta como debilidad o flacidez corporal. Otro rasgo físico frecuentemente observado es la presencia de un único pliegue transversal en la palma de la mano, conocido como pliegue palmar único, que, aunque no es exclusivo del síndrome de Down, es un signo sugestivo cuando se presenta en conjunto con las demás características fenotípicas.
Más allá de los signos morfológicos, existen varias complicaciones médicas que pueden manifestarse al nacimiento o surgir en la infancia temprana. Entre las más importantes se encuentran las malformaciones gastrointestinales, como la atresia duodenal, en la cual existe una interrupción del lumen intestinal que impide el tránsito normal del contenido digestivo. Esta alteración requiere intervención quirúrgica temprana para corregir el defecto anatómico y restaurar la funcionalidad del tracto digestivo. Asimismo, una proporción significativa de neonatos con síndrome de Down presenta cardiopatías congénitas, siendo especialmente comunes los defectos del canal auriculoventricular, que afectan las válvulas y tabiques del corazón. Estos defectos también suelen ser corregibles mediante cirugía cardiovascular especializada.
En el plano hematológico, algunos recién nacidos con síndrome de Down pueden desarrollar una condición llamada leucemia transitoria del recién nacido, caracterizada por la proliferación anormal de blastos en sangre periférica. Esta entidad, aunque alarmante en su presentación inicial, suele resolverse espontáneamente o con un tratamiento conservador. Sin embargo, existe un riesgo significativamente mayor de desarrollar neoplasias hematológicas en etapas posteriores de la infancia, particularmente leucemia linfoblástica aguda y, con mayor frecuencia aún, leucemia mieloide aguda. Estas leucemias tienden a responder a esquemas de quimioterapia ajustados en dosis, aunque el pronóstico empeora considerablemente en casos de recaída.
La capacidad intelectual en individuos con síndrome de Down es muy variable, abarcando desde grados leves hasta moderados de discapacidad cognitiva. Esta variabilidad obedece a múltiples factores genéticos, neurológicos y ambientales. Además, estas personas suelen presentar complicaciones neurológicas y sistémicas adicionales a lo largo de su vida. Entre ellas destacan la inestabilidad atlantoaxoidea —una laxitud entre la primera y segunda vértebras cervicales que puede provocar compresión medular—, deficiencias inmunológicas que aumentan la susceptibilidad a enfermedades autoinmunes como la enfermedad celíaca y la diabetes mellitus tipo 1, infecciones respiratorias recurrentes y disfunción tiroidea, comúnmente en forma de hipotiroidismo.

En términos de desarrollo social y funcional, muchas personas con síndrome de Down logran un buen nivel de integración en entornos protegidos, como talleres ocupacionales y residencias comunitarias supervisadas. Sin embargo, alcanzar una independencia plena en la vida adulta es poco frecuente debido a la combinación de limitaciones cognitivas y complicaciones médicas crónicas. A medida que envejecen, se observa una alta prevalencia de un deterioro cognitivo progresivo que se asemeja a la enfermedad de Alzheimer, con aparición típica entre la cuarta y quinta décadas de vida. Este tipo de demencia está asociado con cambios neuropatológicos similares a los observados en la población general con enfermedad de Alzheimer, incluyendo placas seniles y ovillos neurofibrilares, pero con una aparición más temprana y evolución más acelerada.
Aunque la esperanza de vida de las personas con síndrome de Down ha aumentado notablemente en las últimas décadas gracias a los avances médicos y al mayor acceso a cuidados especializados, aquellos que desarrollan demencia temprana suelen experimentar una reducción significativa en su expectativa de vida. En promedio, los individuos afectados que superan la infancia y llegan a la adultez viven hasta aproximadamente los cincuenta y cinco años.
Exámenes diagnósticos
Aunque la gran mayoría de los casos de síndrome de Down se deben a una trisomía libre del cromosoma 21 —es decir, la presencia de una copia adicional completa de este cromosoma sin ninguna alteración estructural en los demás—, es fundamental realizar un análisis citogenómico completo en todos los pacientes diagnosticados con esta condición. Este estudio no solo permite confirmar el diagnóstico clínico y determinar la causa cromosómica específica del síndrome, sino que también es crucial para identificar casos menos frecuentes en los que el síndrome de Down se origina por una reorganización estructural, en particular una translocación no balanceada.
En algunos individuos afectados, el cromosoma 21 adicional no se encuentra como una copia libre, sino que está fusionado con otro cromosoma a través de una translocación. En estos casos, el paciente hereda una porción extra del material genético del cromosoma 21 que está adherido a otro cromosoma, lo cual provoca un desequilibrio genómico. Este tipo de alteración estructural puede ser el resultado de una translocación desbalanceada que el individuo afectado ha heredado de uno de sus progenitores, quien a su vez puede portar una translocación balanceada. En una translocación balanceada, el material genético total está presente en cantidad normal, pero reorganizado, por lo que el portador suele ser fenotípicamente sano. Sin embargo, durante la formación de gametos, esta reorganización puede dar lugar a combinaciones cromosómicas desequilibradas, aumentando significativamente el riesgo de descendencia con síndrome de Down u otras aneuploidías.
Por esta razón, incluso si el fenotipo del paciente corresponde claramente a una trisomía 21, no es suficiente asumir que se trata de una trisomía libre sin confirmación citogenómica. La identificación de una translocación no solo tiene implicaciones para el paciente, sino también para el asesoramiento genético de la familia. Si se detecta una translocación, es necesario estudiar los cariotipos de ambos padres para determinar si uno de ellos es portador. En caso afirmativo, se reconoce un riesgo de recurrencia considerablemente mayor en futuros embarazos. Además, otros miembros de la familia del portador —como hermanos o hermanas en edad reproductiva— podrían ser también portadores asintomáticos de la misma translocación balanceada y, por tanto, tener un riesgo incrementado de tener hijos con anomalías cromosómicas.
El análisis citogenómico, que incluye técnicas como el cariotipo convencional, la hibridación in situ fluorescente y los estudios con microarreglos cromosómicos, permite detectar tanto aneuploidías como reordenamientos estructurales, ya sean balanceados o desbalanceados. Este tipo de evaluación genética exhaustiva es esencial para brindar una información diagnóstica completa y para establecer un plan de asesoramiento genético adecuado.
Tratamiento
La atresia duodenal, una malformación congénita caracterizada por la interrupción completa o parcial del lumen del duodeno, constituye una urgencia médica que requiere tratamiento quirúrgico. Esta condición impide el paso del contenido gástrico hacia el intestino delgado, lo que provoca obstrucción intestinal alta desde los primeros días de vida. Clínicamente, se manifiesta con vómitos biliosos, distensión abdominal y, en muchos casos, incapacidad para tolerar la alimentación. Si no se corrige oportunamente, la atresia duodenal puede conducir a complicaciones graves, como deshidratación, desequilibrio electrolítico, aspiración pulmonar e incluso perforación intestinal.
El tratamiento definitivo es exclusivamente quirúrgico y consiste en restablecer la continuidad del tracto gastrointestinal mediante técnicas como la duodenoduodenostomía o la duodeno-yeyunostomía, dependiendo del patrón anatómico de la obstrucción. La intervención temprana mejora significativamente el pronóstico y permite un desarrollo gastrointestinal funcional. En pacientes con síndrome de Down, quienes presentan una frecuencia elevada de esta anomalía digestiva, la cirugía se aborda con la misma rigurosidad y técnica que en cualquier otro paciente, aunque con una vigilancia postoperatoria más estrecha debido a la frecuente presencia de comorbilidades.
Entre esas comorbilidades, las cardiopatías congénitas son particularmente relevantes. En los pacientes con síndrome de Down, las malformaciones cardíacas estructurales, como los defectos del canal auriculoventricular, son altamente prevalentes. Estas cardiopatías requieren evaluación exhaustiva por parte de un equipo especializado y deben tratarse siguiendo los mismos principios terapéuticos que se aplican a la población pediátrica general. Dependiendo de la naturaleza y gravedad del defecto, el tratamiento puede incluir manejo farmacológico inicial, seguimiento hemodinámico y, en muchos casos, corrección quirúrgica. El objetivo es preservar la función cardíaca, mejorar la oxigenación y prevenir complicaciones como hipertensión pulmonar y fallo cardíaco.
Por otro lado, en lo que respecta a los aspectos neurológicos del síndrome de Down, tanto el desarrollo cognitivo como los procesos neurodegenerativos posteriores siguen siendo áreas de gran desafío terapéutico. A pesar del considerable interés científico y los múltiples ensayos clínicos realizados, hasta la fecha no se ha demostrado que ningún tratamiento farmacológico o suplementación nutricional pueda mejorar de manera significativa el desarrollo neurocognitivo ni prevenir el deterioro progresivo que suele aparecer en la adultez. Entre las intervenciones exploradas, la administración de antioxidantes como la vitamina E, la vitamina C o la coenzima Q10 ha sido objeto de investigación con la hipótesis de que podrían reducir el estrés oxidativo, el cual se postula como un mecanismo implicado en la neurodegeneración precoz observada en estos pacientes. Sin embargo, los ensayos clínicos disponibles no han mostrado beneficios clínicos concluyentes en términos de función cognitiva, calidad de vida ni progresión de la demencia tipo Alzheimer, común en esta población.
Por tanto, aunque las intervenciones quirúrgicas para las malformaciones gastrointestinales y cardíacas tienen un fundamento terapéutico sólido y una eficacia comprobada, las opciones de tratamiento para las manifestaciones neurológicas del síndrome de Down siguen siendo limitadas. La atención en estos casos debe centrarse en un abordaje multidisciplinario que incluya estimulación temprana, terapias educativas y soporte familiar, en tanto se continúa la búsqueda científica de estrategias que puedan modificar el curso neurológico de esta condición genética.


Fuente y lecturas recomendadas:
-
Boucher AC et al. Clinical and biological aspects of myeloid leukemia in Down syndrome. Leukemia. 2021;35:3352. [PMID: 34518645]
-
Fortea J et al. Alzheimer’s disease associated with Down syndrome: a genetic form of dementia. Lancet Neurol. 2021;20:930. [PMID: 34687637]
-
Gandy KC et al. The relationship between chronic health conditions and cognitive deficits in children, adolescents, and young adults with Down syndrome: a systematic review. PLoS One. 2020;15:e0239040. [PMID: 32915911]
-
Jafri SK et al. Use of antioxidants supplementation of developmental outcomes in children with Down syndrome—a systematic review and meta-analysis. Child Care Health Dev. 2022;48:177. [PMID: 34644809]
-
Shear MA et al. A systematic review and meta-analysis of cell-free DNA testing for detection of fetal sex chromosome aneuploidy. Prenat Diagn. 2023;43:133. [PMID: 36588186]
-
Walpert M et al. A systematic review of unexplained early regression in adolescents and adults with Down syndrome. Brain Sci. 2021;11:1197. [PMID: 34573218]
Aprende administración paso a paso

ADMINISTRACION DESDE CERO

