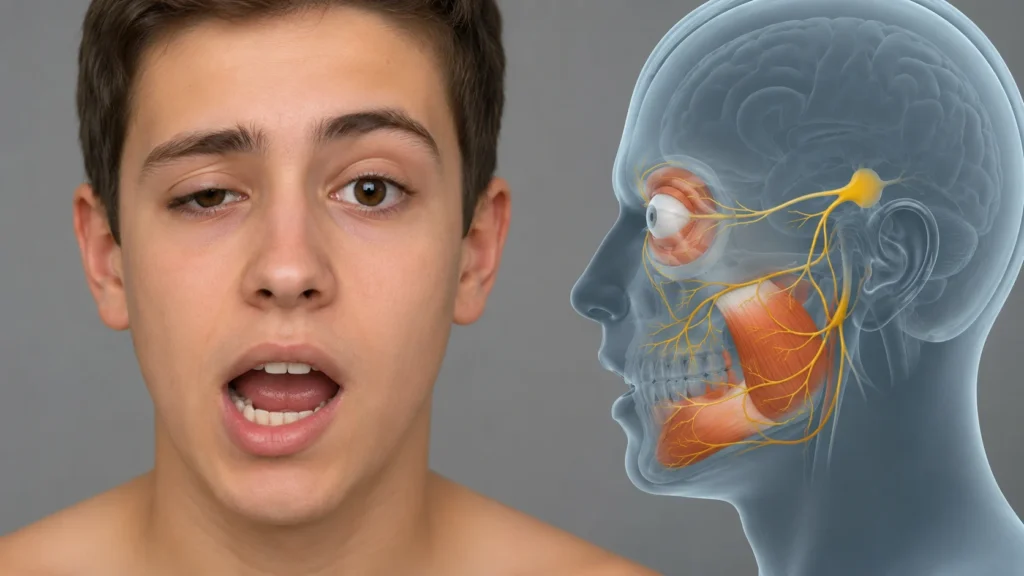
El síndrome de parpadeo de mandíbula de Marcus Gunn, también denominado sincinesia trigemino-oculomotora o sincinesia pterigoideo-elevador, constituye una alteración congénita infrecuente de la inervación craneal caracterizada por un movimiento involuntario del párpado superior asociado de manera constante y reproducible con determinados movimientos mandibulares. El fenómeno consiste en la elevación del párpado afectado durante la apertura de la boca, la masticación, la protrusión mandibular o los desplazamientos laterales de la mandíbula. Este movimiento constituye una sincinesia, es decir, una contracción muscular involuntaria desencadenada por la activación voluntaria de otro grupo muscular anatómica y funcionalmente distinto.
La entidad fue descrita originalmente por Marcus Gunn en 1883 y actualmente se considera una de las formas más representativas de disinervación congénita de los nervios craneales, grupo de enfermedades conocidas como trastornos congénitos de la inervación craneal. Estas enfermedades comparten un mecanismo común basado en alteraciones del desarrollo embrionario de los núcleos motores del tronco encefálico, de sus axones o de sus conexiones periféricas, lo que produce patrones anómalos de activación muscular desde el nacimiento. Esta clasificación explica por qué el síndrome frecuentemente coexiste con otras anomalías oculomotoras congénitas y por qué presenta un comportamiento estable durante toda la vida.
La característica clínica esencial consiste en que el músculo elevador del párpado superior, cuya función fisiológica es abrir el ojo mediante la inervación del nervio oculomotor, se contrae de forma inapropiada cuando se activan músculos de la masticación inervados por la división mandibular del nervio trigémino. Como consecuencia, el paciente presenta una ptosis palpebral en reposo que disminuye parcial o completamente durante determinados movimientos mandibulares.
El control normal del párpado superior depende del núcleo central caudal del complejo oculomotor, localizado en el mesencéfalo. Dicho núcleo envía fibras motoras hacia ambos músculos elevadores del párpado superior. En condiciones normales no existe una conexión funcional entre este núcleo y los núcleos motores del nervio trigémino responsables de la masticación. Por ello, abrir la boca, masticar o desplazar la mandíbula nunca debería modificar la posición del párpado.
En el síndrome de Marcus Gunn esta independencia funcional desaparece debido a una alteración del desarrollo de los circuitos neuronales. El resultado es una activación simultánea del músculo elevador del párpado superior cada vez que determinados músculos masticadores reciben un impulso motor.
La etiología exacta continúa siendo motivo de investigación, aunque existen tres hipótesis fisiopatológicas principales respaldadas por diferentes observaciones clínicas y experimentales.
La primera teoría propone la existencia de conexiones aberrantes localizadas en el mesencéfalo entre el núcleo motor del trigémino, el núcleo mesencefálico del trigémino y el complejo nuclear del nervio oculomotor. Durante el desarrollo embrionario, los axones motores siguen señales moleculares altamente específicas para alcanzar sus músculos diana. Alteraciones discretas en estos mecanismos de guía axonal podrían permitir que algunas fibras destinadas originalmente a músculos masticadores establezcan conexiones inapropiadas con neuronas motoras del músculo elevador del párpado superior. Como consecuencia, un estímulo motor destinado a la mandíbula activaría simultáneamente ambos sistemas musculares.
Esta hipótesis resulta congruente con el carácter congénito de la enfermedad, la ausencia de progresión clínica y la reproducibilidad del movimiento sincinético durante toda la vida.
La segunda hipótesis plantea una reinervación aberrante entre la división mandibular del nervio trigémino, particularmente las fibras destinadas al músculo pterigoideo lateral, y la división superior del nervio oculomotor, responsable de la inervación del músculo elevador del párpado superior. En este escenario existiría un error durante el crecimiento axonal embrionario que permitiría que algunas fibras motoras trigeminales alcanzaran el elevador palpebral. Así, cada vez que el sistema nervioso genera una orden destinada a activar el músculo pterigoideo durante la apertura bucal o la masticación, parte del impulso también llegaría al músculo elevador del párpado, produciendo su contracción involuntaria.
La asociación clínica particularmente intensa entre el parpadeo y la activación del músculo pterigoideo lateral proporciona un importante apoyo anatómico a esta teoría.
Una tercera explicación corresponde a la denominada hipótesis de liberación. Según este modelo, durante etapas tempranas de la evolución filogenética y del desarrollo embrionario existirían reflejos motores compartidos entre la musculatura mandibular y la musculatura palpebral. A medida que madura el sistema nervioso central estos reflejos quedarían inhibidos por circuitos supranucleares. En determinadas circunstancias, alteraciones congénitas del desarrollo del tronco encefálico impedirían dicha inhibición, permitiendo que un reflejo motor normalmente suprimido permanezca funcional durante toda la vida.
Aunque esta teoría explica adecuadamente la naturaleza refleja del fenómeno, actualmente posee menor respaldo anatómico que las hipótesis basadas en conexiones aberrantes.
El desarrollo de las técnicas modernas de resonancia magnética mediante tensor de difusión ha proporcionado evidencia estructural adicional. Diversos estudios han demostrado disminución de la anisotropía fraccional en regiones del tegmento mesencefálico, hallazgo compatible con alteraciones de la organización microestructural de los tractos de sustancia blanca. La reducción de la anisotropía fraccional refleja una menor direccionalidad en la difusión del agua a través de los axones mielinizados, fenómeno que suele indicar alteraciones en la organización de las fibras nerviosas. Estos resultados respaldan la existencia de una anomalía del desarrollo del tronco encefálico y fortalecen la teoría de la disinervación congénita.
El síndrome es raro en la población general, aunque representa una causa relativamente frecuente dentro del grupo de pacientes con ptosis congénita. Se estima que entre el 2 % y el 13 % de los individuos con ptosis congénita presentan esta sincinesia. La amplia variabilidad entre estudios probablemente refleja diferencias metodológicas, criterios diagnósticos y poblaciones analizadas.
En la mayoría de los pacientes el trastorno afecta únicamente un ojo. La presentación unilateral constituye la forma clásica y representa la inmensa mayoría de los casos. La afectación bilateral es excepcional y suele asociarse con alteraciones del desarrollo neurológico más complejas.
La ptosis presente en reposo puede variar desde muy discreta hasta extremadamente severa. Sin embargo, el rasgo definitorio no es el grado de ptosis sino la modificación dinámica de la posición palpebral durante los movimientos mandibulares.
La magnitud del movimiento sincinético suele cuantificarse mediante la excursión vertical del párpado superior. Una excursión inferior a 2 milímetros se considera leve y corresponde aproximadamente al 16 % de los pacientes. La mayoría presenta movimientos entre 2 y 4 milímetros, clasificados como moderados y observados en cerca del 76 % de los casos. Las excursiones iguales o superiores a 5 milímetros se consideran severas y representan aproximadamente el 8 % de los pacientes.
Esta clasificación posee importancia terapéutica porque el grado de excursión determina el impacto estético, funcional y psicológico, además de orientar la estrategia quirúrgica.
El movimiento sincinético suele desencadenarse durante la apertura máxima de la boca, la masticación, la protrusión mandibular, la desviación lateral hacia el lado contralateral y, ocasionalmente, durante la succión o la deglución en lactantes. Todos estos movimientos comparten la activación del músculo pterigoideo lateral, lo cual constituye otro argumento a favor de la hipótesis de la reinervación aberrante.
La enfermedad está presente desde el nacimiento, aunque algunos pacientes no son diagnosticados durante los primeros años de vida porque el fenómeno puede pasar inadvertido hasta que los movimientos mandibulares son más evidentes o hasta que la ptosis comienza a generar preocupación estética.
El síndrome puede asociarse con diversas anomalías oftalmológicas. Una de las más frecuentes es la hipotropía ipsilateral, presente aproximadamente en el 26 % de los pacientes. En esta alteración el ojo afectado se encuentra desplazado hacia abajo respecto al ojo contralateral. Su coexistencia apoya nuevamente el concepto de una alteración más extensa del desarrollo de los circuitos oculomotores.
También pueden observarse estrabismo, limitaciones de la motilidad ocular, anisometropía, errores refractivos y ambliopía. Esta última constituye una de las complicaciones más importantes durante la infancia porque la disminución persistente del estímulo visual puede impedir el desarrollo normal de la corteza visual, originando pérdida permanente de la agudeza visual si no se trata oportunamente.
Por este motivo, la evaluación oftalmológica pediátrica completa representa un componente indispensable del manejo inicial. Debe incluir determinación de la agudeza visual, evaluación de la refracción bajo cicloplejía, estudio de la motilidad ocular y búsqueda sistemática de ambliopía.
El diagnóstico suele establecerse clínicamente mediante la observación directa del fenómeno. Se solicita al paciente abrir ampliamente la boca, desplazar la mandíbula hacia ambos lados, protruir la mandíbula o realizar movimientos de masticación. La elevación reproducible del párpado confirma el diagnóstico en la mayoría de los casos.
Las pruebas de imagen generalmente no son necesarias para confirmar el diagnóstico, aunque pueden resultar útiles cuando existen manifestaciones neurológicas adicionales, dudas diagnósticas o sospecha de otras anomalías estructurales del tronco encefálico.
El tratamiento depende fundamentalmente de la intensidad de la ptosis y del grado de sincinesia.
En pacientes con excursión inferior a 2 milímetros y ptosis mínima puede adoptarse una conducta conservadora basada en observación periódica. Muchos pacientes desarrollan mecanismos compensatorios y utilizan voluntariamente la sincinesia para elevar el párpado durante actividades cotidianas, obteniendo una mejoría funcional suficiente para evitar la cirugía.
Cuando la ptosis genera obstrucción significativa del eje visual, repercusión estética importante o cuando el movimiento sincinético resulta moderado o severo, el tratamiento quirúrgico constituye la mejor alternativa.
La estrategia considerada actualmente como la más eficaz consiste en desactivar completamente el músculo elevador del párpado afectado mediante excisión o desinserción, seguida de suspensión frontal bilateral utilizando el músculo frontal como principal elevador del párpado. El fundamento fisiológico de este procedimiento consiste en eliminar el músculo responsable de la sincinesia y sustituir su función por un mecanismo completamente independiente de la inervación trigeminal.
La suspensión bilateral proporciona una mayor simetría dinámica entre ambos párpados durante la mirada primaria y durante los movimientos oculares, evitando diferencias funcionales que podrían persistir con procedimientos unilaterales.
Las series quirúrgicas publicadas muestran que esta técnica elimina prácticamente el parpadeo mandibular en aproximadamente el 97 % de los pacientes y consigue una diferencia inferior a 1 milímetro entre ambos párpados en cerca del 87 %, resultados que reflejan una elevada eficacia funcional y estética.
Como alternativa, algunos cirujanos realizan suspensión frontal unilateral utilizando cabestrillos de silicona sin resecar el músculo elevador. Este procedimiento reduce considerablemente la intensidad del movimiento sincinético y evita una cirugía más extensa. Sin embargo, aproximadamente el 87 % de los pacientes mantiene un parpadeo residual leve, razón por la cual suele reservarse para casos seleccionados.
Como cualquier procedimiento reconstructivo palpebral, la cirugía puede asociarse con complicaciones. Entre las más frecuentes se encuentran la ptosis de las pestañas y la pérdida del pliegue palpebral superior, cada una observada aproximadamente en el 10 % de los pacientes. También puede desarrollarse entropión en alrededor del 3 %, alteración que provoca la inversión del borde palpebral y el contacto de las pestañas con la superficie corneal, pudiendo producir irritación ocular crónica y lesiones epiteliales.
La planificación preoperatoria requiere una evaluación oftalmológica integral. La ambliopía debe identificarse y tratarse antes de corregir la ptosis porque el éxito funcional depende principalmente de preservar el desarrollo visual. Del mismo modo, cualquier estrabismo vertical significativo debe corregirse previamente, ya que la alineación ocular influye sobre la valoración final de la posición palpebral y sobre el resultado estético de la cirugía.
El síndrome de Marcus Gunn representa un excelente ejemplo de alteración congénita de la organización neuronal del tronco encefálico. La evidencia clínica, anatómica y obtenida mediante neuroimagen respalda que su origen radica en errores del desarrollo de las conexiones entre los sistemas motores trigeminal y oculomotor. Aunque la enfermedad suele ser benigna desde el punto de vista neurológico, puede producir repercusiones visuales, funcionales y psicológicas importantes. El reconocimiento temprano, la búsqueda sistemática de ambliopía y estrabismo, así como la selección adecuada de la técnica quirúrgica, permiten obtener excelentes resultados funcionales y cosméticos en la gran mayoría de los pacientes.

Fuente y lecturas recomendadas:
- Dutton, J. J. (1994). Congenital ptosis. Survey of Ophthalmology, 39(5), 339–376.
- Kanski, J. J., & Bowling, B. (2020). Clinical Ophthalmology: A Systematic Approach (9th ed.). Elsevier.
- Kim, J. W., Kim, S. J., & Lee, S. Y. (2016). Diffusion tensor imaging in congenital cranial dysinnervation disorders. American Journal of Neuroradiology, 37(11), 2144–2150.
- Marcus Gunn, R. (1883). Congenital ptosis with peculiar associated movements of the affected lid. Transactions of the Ophthalmological Society of the United Kingdom, 3, 283–287.
- McNab, A. A. (2001). The Marcus Gunn jaw-winking phenomenon. Journal of Pediatric Ophthalmology and Strabismus, 38(5), 292–296.
- Taylor, D., & Hoyt, C. S. (2017). Pediatric Ophthalmology and Strabismus (5th ed.). Elsevier.
- Wright, K. W., Strube, Y. N. J., & Spiegel, P. H. (2021). Pediatric Ophthalmology and Strabismus (4th ed.). Oxford University Press.
Aprende administración paso a paso

ADMINISTRACION DESDE CERO