El tratamiento con hormona de crecimiento en el contexto del Síndrome de Prader-Willi representa una de las intervenciones endocrinológicas más relevantes y fisiopatológicamente coherentes dentro del manejo integral de esta condición, no únicamente por su efecto sobre la estatura final, sino por su impacto sistémico sobre la composición corporal, el metabolismo energético, el desarrollo neuromotor temprano y el perfil cardiometabólico a largo plazo. Para comprender por qué esta terapia adquiere tal importancia clínica, es necesario integrar la biología del síndrome con la fisiología del eje somatotrópico, así como con la peculiar organización hipotalámica que caracteriza a estos pacientes.
En el síndrome de Prader-Willi existe una alteración genética que afecta la expresión de genes paternos en la región cromosómica quince q once punto dos a q trece, lo cual condiciona una disfunción del hipotálamo. Este centro neuroendocrino, que regula la homeostasis energética, el apetito, el crecimiento y múltiples ejes hormonales, presenta una señalización anómala que explica buena parte del fenotipo clínico: hipotonía neonatal, falla de medro, posteriormente hiperfagia de inicio temprano, disminución del gasto energético basal y alteraciones en la secreción hormonal. Dentro de este contexto, el eje hipotálamo hipofisario somatotrópico suele encontrarse deprimido. De hecho, una proporción muy elevada de pacientes presenta concentraciones disminuidas del factor de crecimiento similar a la insulina tipo uno, lo cual refleja una señal biológica insuficiente de crecimiento, aun cuando la hormona de crecimiento puede estar presente en niveles variables.
La administración exógena de hormona de crecimiento tiene, por tanto, una justificación que trasciende la simple corrección de talla baja. En condiciones fisiológicas, esta hormona actúa estimulando la síntesis proteica, la proliferación celular en cartílago de crecimiento y hueso, y promoviendo la lipólisis. En el síndrome de Prader-Willi, donde existe una tendencia intrínseca a la acumulación de tejido adiposo y una masa muscular reducida, este perfil metabólico resulta particularmente desfavorable, ya que contribuye a la disminución de la movilidad, al empeoramiento de la resistencia a la insulina y a la progresión de comorbilidades cardiometabólicas.
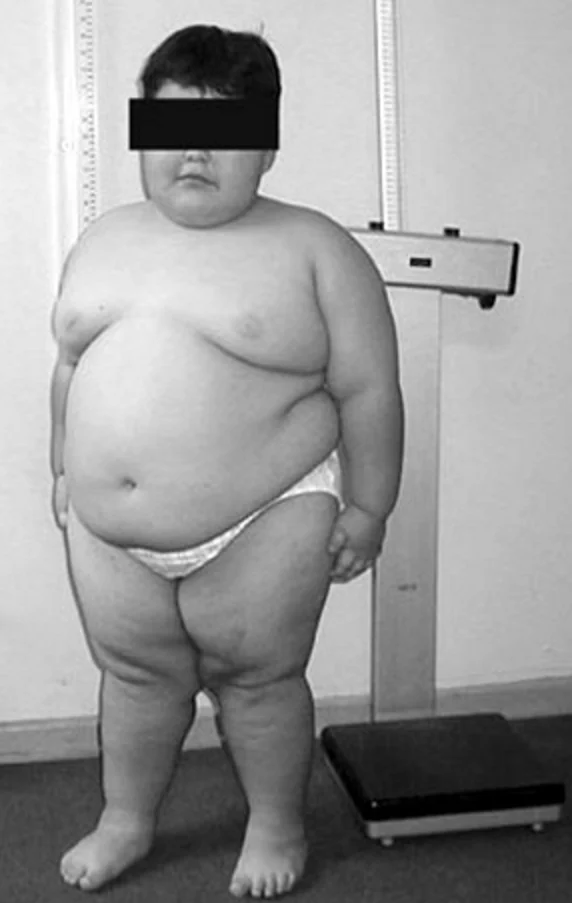
Uno de los efectos más consistentes del tratamiento es la modificación de la composición corporal. Más que un simple incremento de la estatura, la hormona de crecimiento induce un cambio cualitativo en la distribución de tejidos: disminuye la proporción de grasa corporal total, especialmente la grasa visceral, y aumenta la masa magra, incluyendo músculo esquelético. Este efecto es crucial en una población que, incluso desde edades tempranas, presenta un metabolismo energético reducido y una tendencia marcada a la obesidad central. La mejora en la masa muscular no solo tiene implicaciones estéticas o antropométricas, sino funcionales, ya que se traduce en mayor fuerza, mejor control postural y avance más rápido en hitos motores como la sedestación, la bipedestación y la marcha.
Desde el punto de vista del crecimiento lineal, los niños con síndrome de Prader-Willi sin tratamiento suelen alcanzar una talla adulta significativamente inferior a la media poblacional. Esto no se explica únicamente por la alteración hormonal, sino también por la hipotonía crónica y la baja actividad física sostenida. La administración temprana de hormona de crecimiento, especialmente cuando se inicia antes de los dos años de vida, aprovecha una ventana crítica del desarrollo en la que la plasticidad del sistema endocrino y musculoesquelético es mayor. En este periodo, el tratamiento no solo incrementa la velocidad de crecimiento, sino que parece modular de forma más profunda la trayectoria del desarrollo corporal, resultando en una estatura final más cercana a la esperada genéticamente.
Otro aspecto relevante es la influencia de esta terapia sobre el metabolismo lipídico y glucídico. Al aumentar la masa magra y reducir el tejido adiposo, se mejora la sensibilidad a la insulina y se reduce el riesgo de progresión hacia diabetes mellitus tipo dos, una complicación frecuente en estos pacientes debido a la obesidad crónica. Asimismo, se ha observado una mejora en el perfil de colesterol y triglicéridos, lo cual contribuye a disminuir el riesgo cardiovascular a largo plazo. Este efecto metabólico es particularmente importante en un síndrome donde la mortalidad puede estar incrementada por complicaciones asociadas a la obesidad y a la apnea obstructiva del sueño.
La acción de la hormona de crecimiento también se extiende al desarrollo neuromotor. En etapas tempranas de la vida, los pacientes con síndrome de Prader-Willi presentan hipotonía severa que dificulta el movimiento espontáneo y el aprendizaje motor. Al mejorar la masa muscular y la eficiencia neuromuscular, la terapia facilita la adquisición de habilidades motoras y puede contribuir indirectamente al desarrollo cognitivo, al permitir una mayor exploración del entorno y una interacción más rica con estímulos sensoriales.
Sin embargo, el tratamiento no se limita a sus beneficios directos, sino que debe interpretarse dentro de un contexto endocrinológico más amplio. Estos pacientes presentan con mayor frecuencia otras alteraciones del sistema hormonal, como hipotiroidismo central, hipogonadismo hipogonadotrópico e insuficiencia suprarrenal central. Esta última es de particular relevancia clínica, ya que en situaciones de estrés fisiológico, como infecciones graves o cirugías, la incapacidad de aumentar adecuadamente la producción de cortisol puede predisponer a eventos potencialmente fatales. Por ello, el manejo con hormona de crecimiento debe integrarse en una evaluación global del eje endocrino, asegurando la identificación y tratamiento oportuno de estas deficiencias.
El diagnóstico del síndrome se basa en la demostración de una alteración en la expresión génica de origen paterno en la región cromosómica quince q once punto dos a q trece, que puede deberse a deleción, disomía uniparental materna o defectos de impronta. El estudio inicial más sensible es el análisis de metilación del ácido desoxirribonucleico, ya que permite identificar la ausencia de patrón paterno normal independientemente del mecanismo subyacente. Una vez establecido este hallazgo, se utilizan técnicas como la hibridación fluorescente in situ o los microarreglos cromosómicos para precisar la causa estructural específica.

Fuente y lecturas recomendadas:
- Danowitz, M., & Grimberg, A. (2022). Clinical Indications for Growth Hormone Therapy. Advances in pediatrics, 69(1), 203–217. https://doi.org/10.1016/j.yapd.2022.03.005
Aprende administración paso a paso

ADMINISTRACION DESDE CERO