
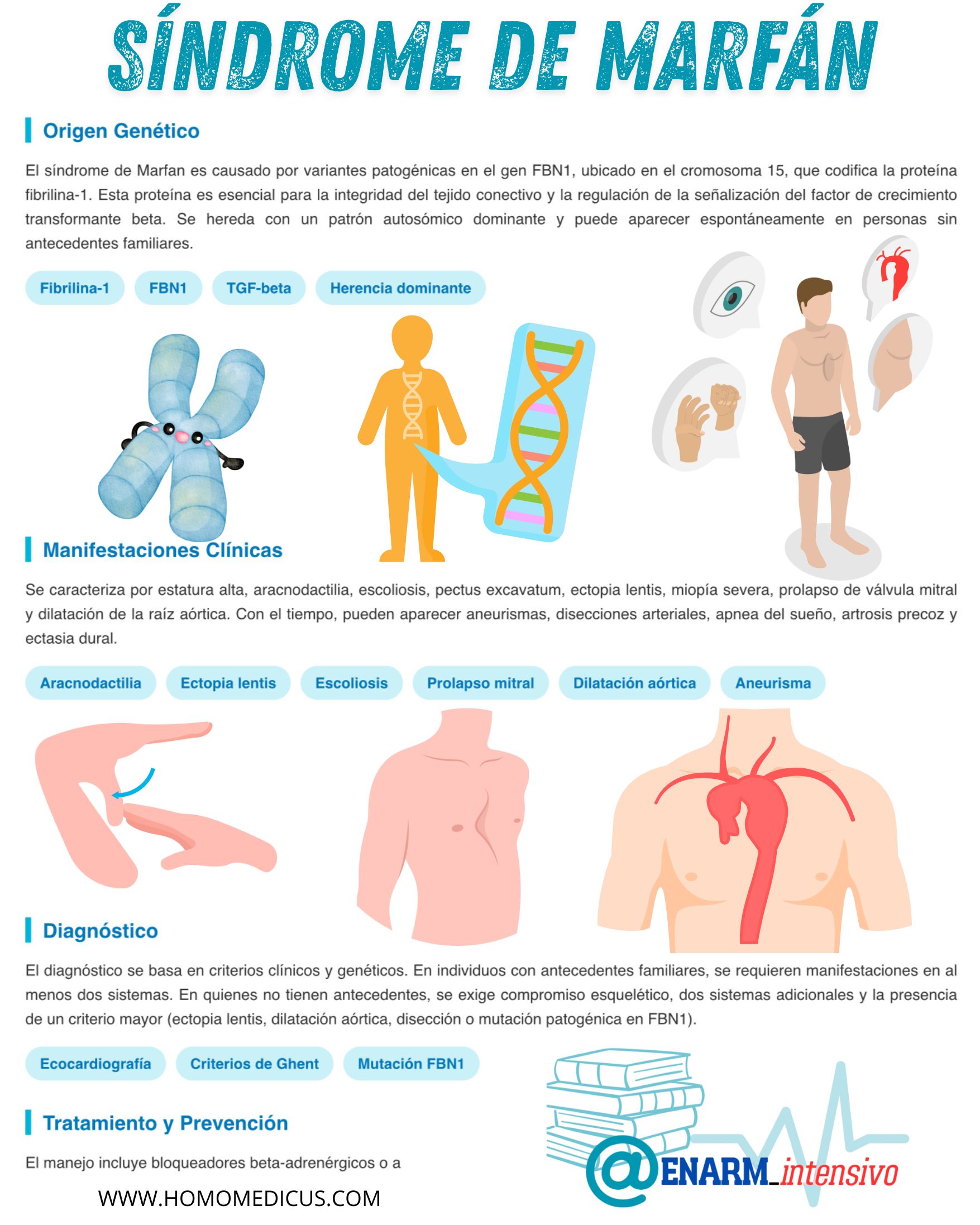
El síndrome de Marfan es una enfermedad sistémica del tejido conectivo que se transmite de manera autosómica dominante. Esta forma de herencia implica que una sola copia alterada del gen responsable es suficiente para causar la enfermedad, incluso si la otra copia es normal. Esto se debe a que el gen implicado, conocido como FBN1, ubicado en el cromosoma 15, codifica una proteína estructural fundamental llamada fibrilina-1, la cual es esencial para la integridad y funcionalidad de las microfibrillas que forman parte del tejido conectivo en múltiples órganos y sistemas.
La alteración genética produce una deficiencia cualitativa o cuantitativa de fibrilina-1, lo que interfiere con la arquitectura del tejido conectivo en todo el organismo. Como resultado, se generan múltiples manifestaciones clínicas, particularmente en los sistemas esquelético, ocular y cardiovascular. Entre las anomalías esqueléticas se encuentran una excesiva longitud de los huesos largos, escoliosis, pectus excavatum o carinatum, y articulaciones hipermóviles. En el sistema ocular, es característica la luxación del cristalino, debido a la debilidad de las zonulas que lo sostienen.
El compromiso cardiovascular, sin embargo, representa el mayor riesgo vital para los pacientes. La raíz de la aorta, que es la porción inicial de este gran vaso sanguíneo, se encuentra especialmente vulnerable. Desde edades tempranas, puede observarse una dilatación progresiva que, en estudios histológicos, revela una degeneración medial difusa. Esta lesión se caracteriza por la fragmentación de las fibras elásticas, la acumulación de mucopolisacáridos y la pérdida de células musculares lisas. Estas alteraciones comprometen la elasticidad y resistencia de la pared aórtica, predisponiendo a aneurismas, disecciones e incluso roturas, eventos potencialmente letales.
Además, el prolapso de la válvula mitral es una manifestación frecuente, resultado de la debilidad del tejido valvular. Esta anomalía puede evolucionar a insuficiencia mitral, contribuyendo a la disfunción cardíaca y a la aparición de síntomas como disnea o palpitaciones.
Otras complicaciones sistémicas incluyen el neumotórax espontáneo, secundario a la ruptura de bullas subpleurales, la ectasia dural (una dilatación del saco dural que puede generar síntomas neurológicos o dolor lumbar crónico), y la aparición de estrías atróficas en la piel, en ausencia de cambios ponderales significativos.
Manifestaciones clínicas
Los pacientes afectados por el síndrome de Marfan presentan, en la mayoría de los casos, una estatura significativamente superior al promedio, acompañada de extremidades desproporcionadamente largas en relación con el tronco. Esta característica se conoce como aracnodactilia, y se manifiesta a través de dedos de manos y pies alargados y delgados, que evocan la apariencia de las patas de una araña. Esta morfología corporal se debe al crecimiento anómalo de los huesos largos, influenciado por alteraciones en la arquitectura del tejido conectivo, específicamente por la deficiencia funcional de la proteína fibrilina-1, implicada en el control del crecimiento y la elasticidad tisular.
A pesar de este fenotipo característico, la expresión clínica del síndrome es sumamente variable entre individuos, incluso dentro de una misma familia. Esta heterogeneidad se traduce en que algunas personas pueden presentar únicamente rasgos esqueléticos leves, mientras que otras desarrollan complicaciones multiorgánicas de gran severidad. Entre las alteraciones musculoesqueléticas más frecuentes se encuentran la escoliosis toracolumbar de evolución progresiva y las deformidades del tórax anterior, como el pectus excavatum (hundimiento del esternón) o el pectus carinatum (proyección anterior del mismo).
El sistema ocular también se ve comprometido, con la luxación del cristalino (ectopia lentis) presente en aproximadamente la mitad de los casos. Esta condición, resultado de la debilidad de las fibras zonulares que mantienen al cristalino en su lugar, puede ocasionar visión doble, desenfoque visual y predisposición a otras complicaciones oculares. La miopía severa es común, lo que aumenta el riesgo de desprendimiento de retina, una emergencia oftalmológica potencialmente catastrófica.
En el aparato cardiovascular, más del cincuenta por ciento de los pacientes desarrolla prolapso de la válvula mitral, condición en la que los velos de la válvula se desplazan de manera anómala hacia la aurícula izquierda durante la sístole, generando insuficiencia mitral. No obstante, la complicación cardiovascular más grave es la dilatación de la raíz aórtica, un hallazgo frecuente que puede evolucionar hacia insuficiencia aórtica, disección o ruptura de la aorta. Estudios recientes han revelado que los aneurismas también pueden desarrollarse en las arterias derivadas de la aorta, como las arterias subclavia o carótida, lo cual anteriormente era subestimado.
El diagnóstico del síndrome de Marfan se establece mediante criterios clínicos definidos por sistemas afectados y antecedentes familiares. En personas con un familiar diagnosticado, es necesario demostrar el compromiso de al menos dos sistemas orgánicos. En cambio, en individuos sin antecedentes familiares conocidos, el diagnóstico requiere evidencia de afectación esquelética, alteraciones en al menos dos sistemas adicionales y la presencia de uno de los siguientes criterios mayores: luxación del cristalino, dilatación de la raíz aórtica, disección aórtica o la identificación de una variante patogénica confirmada en el gen de la fibrilina-1.
Exámenes diagnósticos
El síndrome de Marfan es causado por variantes patogénicas en el gen de la fibrilina-1, localizado en el brazo largo del cromosoma 15. Este gen codifica una glicoproteína estructural fundamental en la formación de las microfibrillas del tejido conectivo. Las microfibrillas no solo proporcionan soporte mecánico a diversos tejidos, sino que también cumplen funciones reguladoras, en particular en la modulación de señales bioquímicas extracelulares. La alteración de este componente básico compromete la arquitectura y la función del tejido conectivo en múltiples órganos.
A pesar del conocimiento detallado de la mutación genética subyacente, el diagnóstico del síndrome de Marfan no puede basarse exclusivamente en pruebas de laboratorio. Esto se debe a que las mutaciones en el gen de la fibrilina también están implicadas en otros síndromes del espectro del tejido conectivo, algunos de los cuales pueden presentar manifestaciones clínicas similares, como la dilatación de la raíz aórtica o el prolapso valvular. Por tanto, la identificación de una variante genética en FBN1, por sí sola, no es concluyente en todos los casos. La evaluación clínica, junto con la historia familiar y los hallazgos en diversos sistemas, sigue siendo esencial para establecer un diagnóstico definitivo.
La patogenia del síndrome de Marfan no se limita únicamente al daño estructural del tejido conectivo. Se ha demostrado que las alteraciones en fibrilina-1 afectan profundamente la regulación de la señalización del factor de crecimiento transformante beta, una molécula con funciones clave en el desarrollo, la reparación tisular y la homeostasis vascular. En condiciones normales, la fibrilina-1 ayuda a secuestrar el factor de crecimiento transformante beta en la matriz extracelular, limitando su actividad biológica. Cuando esta función reguladora se pierde por mutaciones en FBN1, se produce una activación descontrolada de esta vía, lo que contribuye a procesos patológicos como la degradación de la matriz, el debilitamiento de la pared aórtica y la progresión de los aneurismas.
Además, mutaciones en los genes que codifican los receptores del factor de crecimiento transformante beta, concretamente TGFBR1 y TGFBR2, también pueden causar síndromes clínicamente similares al síndrome de Marfan, como el síndrome de Loeys-Dietz. Estas condiciones comparten características clave, como la dilatación y disección de la aorta torácica, así como un patrón de herencia autosómico dominante. Esto subraya el papel central de la vía del factor de crecimiento transformante beta en la fisiopatología de los trastornos del tejido conectivo, más allá de las mutaciones exclusivamente en FBN1.
Por otra parte, se ha identificado un número creciente de genes—más de dos docenas—cuyas variantes pueden predisponer a los adultos al desarrollo de aneurismas y disecciones de la aorta torácica. Esta diversidad genética evidencia que el fenotipo aórtico no es exclusivo del síndrome de Marfan, y que la susceptibilidad a complicaciones vasculares puede tener múltiples orígenes moleculares, algunos aún en proceso de caracterización.
Prevención
La identificación precisa de una variante patogénica en el gen de la fibrilina-1 permite implementar estrategias de prevención genética en familias afectadas por el síndrome de Marfan. Una vez que se ha caracterizado la mutación específica responsable de la enfermedad en un individuo diagnosticado, es posible realizar un diagnóstico genético tanto prenatal como presintomático en familiares en riesgo. Este enfoque se basa en la posibilidad de detectar la mutación conocida mediante técnicas moleculares avanzadas en etapas tempranas del desarrollo embrionario o en individuos que aún no han manifestado signos clínicos.
En el contexto prenatal, se puede obtener material genético fetal a través de procedimientos como la biopsia de vellosidades coriónicas o la amniocentesis. Estas muestras permiten analizar el ADN fetal y determinar si el embrión ha heredado la variante patogénica previamente identificada en el progenitor afectado. Esta información puede ser utilizada por los padres para tomar decisiones informadas sobre el curso del embarazo, especialmente en aquellos casos en que la historia familiar incluya formas graves o de rápida progresión de la enfermedad.
Por otro lado, el diagnóstico presintomático se dirige a individuos nacidos que pertenecen a familias con antecedentes de síndrome de Marfan confirmado por análisis molecular. Estos individuos pueden someterse a pruebas genéticas para determinar si portan la mutación, incluso antes de que se manifiesten signos clínicos. La identificación precoz de personas portadoras posibilita una vigilancia médica estrecha desde edades tempranas, lo que permite intervenir de manera oportuna ante las primeras señales de compromiso cardiovascular u ocular, reduciendo así el riesgo de complicaciones graves.
Además de su utilidad en la prevención clínica, el conocimiento de la mutación específica también resulta fundamental en el asesoramiento genético. Las parejas con antecedentes familiares de síndrome de Marfan pueden recibir orientación sobre el riesgo de transmisión a su descendencia y sobre las alternativas reproductivas disponibles, como el diagnóstico genético preimplantacional. Esta técnica permite seleccionar embriones libres de la mutación durante procedimientos de fertilización in vitro, lo que representa una opción valiosa para evitar la transmisión del trastorno a las generaciones futuras.
Tratamiento
Los niños diagnosticados con síndrome de Marfan requieren una vigilancia médica sistemática y multidisciplinaria desde una edad temprana, con el objetivo de prevenir complicaciones irreversibles y mejorar su pronóstico a largo plazo. La implicación ocular, en particular la luxación del cristalino o ectopia lentis, es una manifestación frecuente de esta enfermedad del tejido conectivo. Esta alteración puede interferir significativamente con el desarrollo normal de la agudeza visual en la infancia, lo que, de no detectarse y corregirse de manera oportuna, puede dar lugar a ambliopía, una forma de pérdida visual permanente por falta de estimulación adecuada del sistema visual en etapas críticas del desarrollo. Por ello, es fundamental realizar evaluaciones oftalmológicas periódicas, que permitan identificar precozmente la ectopia lentis, corregir los defectos refractivos asociados y preservar la función visual.
Asimismo, el compromiso musculoesquelético progresivo, especialmente la escoliosis, requiere seguimiento ortopédico anual durante el crecimiento. La detección temprana de desviaciones en la columna vertebral permite implementar medidas ortésicas, como el uso de corsés, que pueden retrasar la progresión de la curvatura y evitar, en muchos casos, la necesidad de una intervención quirúrgica compleja. Este enfoque es particularmente importante durante la adolescencia, cuando se produce un rápido crecimiento corporal que puede agravar las deformidades estructurales.
A nivel cardiovascular, la evaluación ecocardiográfica anual es indispensable en pacientes de todas las edades. Esta técnica permite monitorear con precisión el diámetro de la raíz aórtica, cuya dilatación progresiva es una de las principales causas de mortalidad en el síndrome de Marfan, así como valorar la función de la válvula mitral, que frecuentemente presenta prolapso e insuficiencia. El seguimiento ecográfico permite detectar cambios estructurales antes de que se manifiesten clínicamente y así guiar decisiones terapéuticas preventivas.
En cuanto al tratamiento farmacológico, se ha demostrado que el bloqueo beta-adrenérgico prolongado, ajustado a la tolerancia individual, tiene un efecto protector sobre la aorta al reducir la fuerza de contracción cardíaca y la presión sobre la pared vascular. Fármacos como el atenolol, administrado en dosis que logren un efecto inotrópico negativo, disminuyen la velocidad de crecimiento de la raíz aórtica. Los antagonistas del receptor de angiotensina, como el losartán, han mostrado una eficacia comparable, probablemente debido a su capacidad para modular la señalización del factor de crecimiento transformante beta, implicado en la patogénesis de la enfermedad. Por el contrario, los bloqueadores de los canales de calcio, anteriormente utilizados como alternativa, han demostrado ser perjudiciales para la integridad de la aorta y, por tanto, se desaconseja su uso en este contexto.
Las restricciones físicas también juegan un papel preventivo importante. Si bien el ejercicio físico moderado no está contraindicado, la participación en actividades de alta intensidad o deportes de contacto debe evitarse, dado que estos incrementan el riesgo de disección aórtica por sobrecarga hemodinámica. El control estricto de estos factores es crucial para preservar la integridad vascular en pacientes con predisposición genética a aneurismas y rupturas.
Cuando el diámetro de la raíz aórtica alcanza entre 45 y 50 milímetros en adultos —un valor superior al rango normal, que suele ser menor a 40 milímetros— se recomienda la cirugía profiláctica para reemplazar la porción afectada. Este procedimiento ha demostrado aumentar significativamente la esperanza de vida al prevenir complicaciones catastróficas. En ciertos casos, como aquellos con antecedentes familiares de disección aórtica o en quienes se observa una tasa de crecimiento acelerada del diámetro aórtico (mayor a 3 o 4 milímetros por año), la intervención quirúrgica debe considerarse de forma anticipada.
Una de las técnicas quirúrgicas más utilizadas es el procedimiento de preservación valvular, en el cual se mantiene la válvula aórtica nativa del paciente y se implanta una prótesis tubular que sustituye las porciones aneurismáticas del seno de Valsalva y de la aorta ascendente. Esta técnica permite evitar la necesidad de anticoagulación crónica, preservando la función valvular natural y reduciendo los riesgos asociados al uso prolongado de anticoagulantes.
Por último, se debe prestar especial atención a las mujeres con síndrome de Marfan que deseen embarazarse. El periodo periparto y posparto representa un momento de alto riesgo para disección aórtica debido a los cambios hemodinámicos y hormonales asociados al embarazo. Por esta razón, en mujeres cuya raíz aórtica exceda los 40 milímetros, debe contemplarse la opción de una cirugía profiláctica de reparación aórtica antes de intentar una gestación. Este enfoque preventivo puede reducir significativamente la morbimortalidad materna durante esta etapa crítica.
Pronóstico
El pronóstico de las personas con síndrome de Marfan ha experimentado una transformación significativa en las últimas décadas, principalmente gracias a los avances en el diagnóstico precoz, el tratamiento médico preventivo y la intervención quirúrgica oportuna. En ausencia de tratamiento, los individuos afectados solían fallecer en la cuarta o quinta década de la vida, siendo la principal causa la disección aórtica aguda o la insuficiencia cardíaca congestiva secundaria a la regurgitación crónica de las válvulas aórtica o mitral. Estas complicaciones cardiovasculares, consecuencia directa del debilitamiento progresivo del tejido conectivo en la raíz aórtica y las estructuras valvulares, representaban históricamente el principal obstáculo para la longevidad en estos pacientes.
Actualmente, el panorama es considerablemente más favorable. La detección temprana, facilitada por la concienciación clínica y el uso de criterios diagnósticos más precisos, ha permitido instaurar medidas de prevención desde edades tempranas. El tratamiento farmacológico con bloqueadores beta-adrenérgicos o antagonistas del receptor de angiotensina ha demostrado ser eficaz para reducir la velocidad de dilatación de la raíz aórtica, disminuyendo así el riesgo de disección. A esto se suma la posibilidad de realizar cirugías profilácticas tanto sobre la raíz aórtica como sobre la válvula mitral, antes de que aparezcan complicaciones graves. Estas intervenciones quirúrgicas, especialmente aquellas que preservan las válvulas nativas, han demostrado mejorar significativamente la calidad y la expectativa de vida, que ahora puede extenderse hasta la séptima u octava década en pacientes adecuadamente tratados.
Sin embargo, este aumento en la longevidad ha traído consigo un fenómeno clínico emergente: la aparición de comorbilidades que anteriormente eran poco frecuentes o pasaban desapercibidas debido a la menor esperanza de vida. Entre estas se encuentra la apnea obstructiva del sueño, una condición respiratoria que puede exacerbar la hipertensión y la disfunción cardiovascular. Asimismo, algunos pacientes desarrollan miocardiopatía, una alteración del músculo cardíaco que puede comprometer la función sistólica o diastólica con el tiempo.
También se ha documentado un aumento en la incidencia de aneurismas y disecciones no solo en la aorta torácica, sino también en la aorta abdominal y en las arterias periféricas, lo cual requiere ampliar la vigilancia vascular más allá del eje aórtico central. En el sistema nervioso, las consecuencias de la ectasia dural, como el dolor lumbar crónico, alteraciones sensitivas o disfunción de raíces nerviosas, pueden volverse cada vez más relevantes con el paso del tiempo. Por último, los trastornos osteoarticulares degenerativos, como la artrosis prematura, también se presentan con mayor frecuencia, posiblemente como consecuencia de la hiperlaxitud articular crónica y del desgaste mecánico acumulado a lo largo de los años.
En resumen, el pronóstico de los pacientes con síndrome de Marfan ha mejorado sustancialmente gracias a un enfoque preventivo integral que combina la vigilancia clínica rigurosa, el tratamiento farmacológico dirigido y la cirugía profiláctica. No obstante, esta mayor sobrevida plantea nuevos desafíos clínicos relacionados con la aparición de comorbilidades propias de una vida más prolongada en un contexto de fragilidad estructural generalizada. Este cambio obliga a replantear el seguimiento a largo plazo y a incorporar estrategias multidisciplinarias que aborden tanto las complicaciones cardiovasculares como los efectos multisistémicos de la enfermedad.

Fuente y lecturas recomendadas:
- Goldman, L., & Schafer, A. I. (Eds.). (2020). Goldman-Cecil Medicine (26th ed.). Elsevier.
- Loscalzo, J., Fauci, A. S., Kasper, D. L., Hauser, S. L., Longo, D. L., & Jameson, J. L. (Eds.). (2022). Harrison. Principios de medicina interna (21.ª ed.). McGraw-Hill Education.
- Papadakis, M. A., McPhee, S. J., Rabow, M. W., & McQuaid, K. R. (Eds.). (2024). Diagnóstico clínico y tratamiento 2025. McGraw Hill.
- Rozman, C., & Cardellach López, F. (Eds.). (2024). Medicina interna (20.ª ed.). Elsevier España.
Aprende administración paso a paso

ADMINISTRACION DESDE CERO

