
El síndrome de von Willebrand (VWD) es una afección hereditaria que afecta la coagulación sanguínea debido a una deficiencia o disfunción en la proteína von Willebrand (vWF), que desempeña un papel crucial en el proceso de coagulación. Hay varios tipos de enfermedad de von Willebrand, que se clasifican según la gravedad de la deficiencia de vWF y la naturaleza específica de la disfunción. A continuación, se describen los tres tipos principales:
- Tipo 1: Este es el tipo más común y se caracteriza por una deficiencia parcial de la proteína vWF. Los pacientes con tipo 1 suelen tener niveles reducidos de vWF en su torrente sanguíneo, pero la proteína que producen es funcional. Los síntomas pueden variar desde leves hasta moderados, y los pacientes pueden experimentar sangrado prolongado después de lesiones menores o cirugías.
- Tipo 2: En este tipo, la proteína vWF está presente en cantidades normales, pero es defectuosa en su estructura o función. Se han identificado varios subtipos dentro de la clasificación del tipo 2, según la naturaleza específica de la anomalía en la vWF. Estos subtipos incluyen:
- Tipo 2A: Se caracteriza por una vWF que se degrada más rápidamente de lo normal.
- Tipo 2B: La vWF tiene una mayor afinidad por las plaquetas, lo que puede llevar a una trombocitopenia (niveles bajos de plaquetas) y un mayor riesgo de formación de coágulos.
- Tipo 2M: La vWF tiene una disminución en su capacidad para unirse a las plaquetas.
- Tipo 2N: Hay una disminución en la capacidad de la vWF para unirse al factor VIII, lo que resulta en niveles bajos de factor VIII en la sangre.
- Tipo 3: Este es el tipo más grave y raro de VWD, caracterizado por una deficiencia completa o casi completa de vWF en la sangre. Los pacientes con tipo 3 tienen un alto riesgo de sangrado severo, que puede ocurrir espontáneamente o después de lesiones menores. Este tipo de VWD se hereda de manera autosómica recesiva, lo que significa que ambos padres deben transmitir el gen defectuoso para que un individuo desarrolle la enfermedad.
Además de estos tipos principales, también se han descrito variantes y subtipos menos comunes de la enfermedad de von Willebrand, lo que refleja la complejidad y diversidad de esta condición hematológica.
Factor de von Willebrand
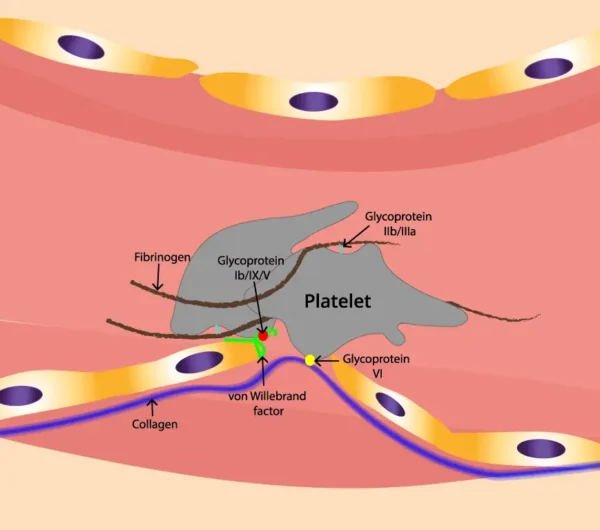
El factor de von Willebrand (vWF) es una glucoproteína esencial en la cascada de coagulación sanguínea, cuya función principal radica en el proceso de hemostasia primaria. Su naturaleza multímera y su capacidad para interactuar con diversas moléculas y componentes del entorno vascular lo convierten en un actor crucial en la formación del tapón plaquetario, un evento fundamental para detener la hemorragia en caso de lesión vascular.
La peculiaridad del vWF reside en su estructura y en sus múltiples funciones dentro del sistema hemostático. En primer lugar, destaca su tamaño inusualmente grande, que se debe a la presencia de múltiples dominios de repetición de tipo A (dominios A) y un dominio de unión al colágeno tipo C (dominio C). Estos dominios son esenciales para su capacidad de unirse tanto al colágeno subendotelial como a la glicoproteína Ib (GPIb), presente en la membrana de las plaquetas.
La interacción del vWF con el colágeno subendotelial es crucial para la adhesión plaquetaria en el sitio de lesión vascular. El vWF se une al colágeno expuesto en la lámina subendotelial después de una lesión vascular, formando un puente entre las plaquetas circulantes y la matriz extracelular dañada. Esta unión mediada por el vWF proporciona un anclaje firme para las plaquetas en el sitio de la lesión, facilitando así la formación del tapón plaquetario.
Además de su interacción con el colágeno, el vWF también se une a la GPIb presente en la membrana plaquetaria. Esta interacción es esencial para la unión inicial de las plaquetas al vaso sanguíneo lesionado. La unión del vWF a la GPIb en la superficie de las plaquetas activadas no solo contribuye a la adhesión plaquetaria, sino que también desencadena la activación plaquetaria, lo que lleva a la liberación de más vWF y al cambio conformacional de la GPIIb/IIIa, facilitando la agregación plaquetaria.
Características de los tipos de enfermedad de von Willebrand
El factor de von Willebrand (vWF) tiene un sitio de unión específico para el factor VIII (FVIII) en la circulación sanguínea. Esta interacción permite que el vWF actúe como un portador o transportador del FVIII, prolongando su vida media en la circulación. Esta asociación entre el vWF y el FVIII es esencial para mantener niveles adecuados de FVIII, que a su vez es crucial para la formación de trombina y la hemostasia efectiva. En la enfermedad de von Willebrand, especialmente en el tipo 1, que es la forma más común, hay una anormalidad cuantitativa en la molécula de vWF, lo que afecta su capacidad para unirse al FVIII. Aunque la causa exacta del tipo 1 de la enfermedad de von Willebrand no siempre se identifica, esta anormalidad en la cantidad de vWF puede provocar una disminución en la estabilidad y la función del FVIII, lo que contribuye a los síntomas de sangrado observados en estos pacientes.
El tipo 2 de la enfermedad de von Willebrand (vWD) afecta al 15-20% de los pacientes con esta condición y se subdivide en varios subtipos, cada uno con características distintivas. En los tipos 2A o 2B de vWD, se observan defectos cualitativos en la molécula de factor de von Willebrand (vWF), lo que compromete su capacidad para funcionar correctamente. Por otro lado, los tipos 2N y 2M de vWD se deben a defectos específicos en el vWF que afectan su unión al factor VIII o a las plaquetas, respectivamente.
El tipo 2M de vWD se caracteriza por un patrón multímero normal del vWF, pero con una disminución en su capacidad para unirse a las plaquetas. Por otro lado, el tipo 2N de vWD puede presentar síntomas clínicos similares a la hemofilia A debido a que los niveles de actividad del factor VIII están disminuidos, a pesar de que la actividad y el antígeno del vWF son normales. Esta similitud clínica puede conducir a un diagnóstico erróneo, lo que subraya la importancia de una evaluación cuidadosa para distinguir entre estas condiciones.
El tipo 3 de enfermedad de von Willebrand (vWD) es una forma rara de la enfermedad que, al igual que el tipo 1, se caracteriza por un defecto cuantitativo en la molécula de factor de von Willebrand (vWF). En este tipo de vWD, los pacientes presentan niveles muy bajos o incluso ausentes de vWF en la circulación sanguínea, lo que resulta en un riesgo significativo de sangrado grave desde la infancia o la niñez.
Dado que el vWF actúa como un portador de factor VIII (FVIII) en la circulación, la deficiencia grave de vWF también conduce a una baja actividad del FVIII. Esta deficiencia de FVIII contribuye aún más al riesgo de sangrado al afectar la capacidad de formación del coágulo. Un tiempo prolongado de tromboplastina parcial activada (aPTT) es una característica común en estos pacientes, lo que refleja la disminución en la actividad del FVIII y la alteración en la cascada de coagulación.

Aprende administración paso a paso

ADMINISTRACION DESDE CERO

