La poliangeítis microscópica es una forma de vasculitis necrotizante no granulomatosa y pauci-inmune, lo que significa que se caracteriza por la inflamación y destrucción de los vasos sanguíneos pequeños, sin la presencia de granulomas típicos que se observan en otras formas de vasculitis. Esta enfermedad afecta principalmente a los vasos sanguíneos pequeños, como los capilares, las vénulas y las arteriolas, que son estructuras clave en la circulación sanguínea de los órganos y tejidos. En consecuencia, la poliangeítis microscópica puede provocar alteraciones significativas en la perfusión y el suministro de oxígeno y nutrientes a las células de los órganos afectados, lo que a su vez desencadena una serie de respuestas inflamatorias y daños tisulares.
Uno de los hallazgos más comunes en los pacientes con poliangeítis microscópica es la glomerulonefritis, que es una inflamación de los glomérulos renales. Los glomérulos son estructuras microscópicas dentro de los riñones que desempeñan un papel fundamental en la filtración de la sangre. Cuando se inflaman, pueden causar una disminución de la función renal, lo que puede llevar a insuficiencia renal si no se trata adecuadamente. Además, la poliangeítis microscópica también se asocia frecuentemente con la capilritis pulmonar, que es la inflamación de los capilares en los pulmones, lo que puede causar hemorragias pulmonares y dificultades respiratorias.
El diagnóstico de esta enfermedad se ve facilitado por la presencia de anticuerpos anticitoplasma de neutrófilos (ANCA, por sus siglas en inglés). En particular, la forma de ANCA denominada p-ANCA es frecuente en los pacientes con poliangeítis microscópica. Estos anticuerpos atacan los neutrófilos, que son células clave del sistema inmunológico, y pueden desencadenar una respuesta inflamatoria en los vasos sanguíneos, lo que perpetúa el daño y la necrosis de los vasos afectados.
A pesar de ser principalmente una vasculitis de los vasos pequeños, la poliangeítis microscópica puede involucrar vasos sanguíneos de tamaño medio, lo que puede superponerse con las características clínicas de otras vasculitis, como la poliarteritis nodosa y la granulomatosis con poliangeítis. La poliarteritis nodosa afecta típicamente a arterias de mayor calibre, mientras que la granulomatosis con poliangeítis también involucra la formación de granulomas, una característica distintiva de esa enfermedad. Sin embargo, la poliangeítis microscópica comparte con ambas condiciones la propensión a afectar a los riñones y los pulmones, lo que genera un solapamiento en el espectro clínico de estas enfermedades vasculares.
En algunos casos raros, ciertos medicamentos pueden inducir una vasculitis sistémica que presenta características similares a las de la poliangeítis microscópica. Medicamentos como el propiltiouracilo, la hidralazina, el alopurinol, la penicilamina, la minociclina y la sulfasalazina se han relacionado con el desarrollo de esta vasculitis. Estos fármacos pueden desencadenar una reacción inmunológica en el organismo, que conduce a la producción de p-ANCA y provoca inflamación y daño en los vasos sanguíneos, lo que en ocasiones se manifiesta con los mismos síntomas clínicos de la poliangeítis microscópica. Sin embargo, este tipo de vasculitis inducida por medicamentos es relativamente poco frecuente y generalmente se resuelve al interrumpir el uso del fármaco causante.
Manifestaciones clínicas
La poliangeítis microscópica es una forma de vasculitis que afecta principalmente a los vasos sanguíneos pequeños y puede dar lugar a una amplia variedad de hallazgos clínicos que sugieren la presencia de vasculitis en dichos vasos. Estos hallazgos incluyen la aparición de púrpura palpable o elevada, que es una manifestación común de vasculitis cutánea. La púrpura palpable se caracteriza por manchas violáceas o rojizas en la piel, que resultan de la extravasación de sangre debido a la inflamación de los vasos sanguíneos pequeños. Además de la púrpura, pueden desarrollarse otros signos de vasculitis cutánea, como úlceras, hemorragias en astilla (pequeñas hemorragias bajo las uñas) y lesiones vesiculobullosas, que son ampollas o vesículas llenas de líquido que pueden aparecer en la piel o las mucosas.
Uno de los aspectos más relevantes de la poliangeítis microscópica es su relación con el síndrome pulmonar-renal, que es un conjunto de síntomas que implica tanto a los pulmones como a los riñones. Este síndrome es causado con mayor frecuencia por la poliangeítis microscópica que por otras enfermedades, como la enfermedad de la membrana basal glomerular anti-glomerular. En este contexto, la poliangeítis microscópica es la causa más común de insuficiencia renal asociada a hemorragias pulmonares. La hemorragia pulmonar ocurre debido a la inflamación de los capilares pulmonares, lo que provoca la ruptura de los vasos sanguíneos y la extravasación de sangre hacia los pulmones. Histológicamente, se observan cambios típicos de capilritis, que es la inflamación de los capilares, un hallazgo común en la poliangeítis microscópica.
Además de las hemorragias pulmonares, la poliangeítis microscópica puede presentar fibrosis intersticial en los pulmones, una condición en la cual el tejido pulmonar se vuelve más rígido debido a la formación de tejido cicatricial. Esta fibrosis puede simular la neumonitis intersticial usual, una enfermedad pulmonar progresiva que también se asocia con un mal pronóstico. La presencia de fibrosis intersticial en los pulmones puede afectar gravemente la función respiratoria y complicar el pronóstico general de los pacientes.
Otro hallazgo común en los pacientes con poliangeítis microscópica es la neuropatía vasculítica, que se presenta con mayor frecuencia como una mononeuritis múltiple. La mononeuritis múltiple es una condición en la cual varios nervios periféricos se inflaman de manera aislada, lo que puede causar dolor, debilidad, entumecimiento y pérdida de función en las áreas afectadas. Esta neuropatía es consecuencia de la inflamación de los vasos sanguíneos que irrigan los nervios, lo que interfiere con el suministro de sangre y oxígeno a los nervios periféricos, y puede llevar a un daño nervioso significativo.
Exámenes diagnósticos
En los estudios de laboratorio, uno de los hallazgos más relevantes es la presencia de anticuerpos anticitoplasma de neutrófilos (ANCA, por sus siglas en inglés), que se observan en la mayoría de los casos de esta enfermedad.
En aproximadamente tres cuartas partes de los pacientes con granulomatosis eosinofílica con poliangeítis, se detectan anticuerpos ANCA en suero, predominantemente en forma de anticuerpos dirigidos contra la mieloperoxidasa (anti-MPO-ANCA). Estos anticuerpos son responsables de inducir una reacción inmune que genera una respuesta inflamatoria en los vasos sanguíneos. En los estudios de inmunofluorescencia, los anticuerpos anti-MPO-ANCA generalmente provocan un patrón llamado p-ANCA, que es característico de la enfermedad. En menor frecuencia, también pueden encontrarse anticuerpos dirigidos contra la proteínaasa-3 (PR3-ANCA), otro tipo de ANCA que se observa en casos específicos de vasculitis.
Además de la presencia de los ANCA, otro hallazgo clave en los pacientes con esta enfermedad es la elevación de los reactantes de fase aguda, que son proteínas plasmáticas cuya concentración aumenta en respuesta a la inflamación. Los reactantes más comunes que se elevan son la proteína C reactiva y la velocidad de sedimentación de los eritrocitos. Estas elevaciones reflejan la actividad inflamatoria y la respuesta inmune del organismo frente al daño vascular y tisular que ocurre en la enfermedad.
En cuanto a la afectación renal, es frecuente la presencia de hematuria microscópica, que se detecta mediante el análisis de orina, además de proteinuria. Estos hallazgos indican daño en los glomérulos, las estructuras renales responsables de la filtración de sangre. En algunos casos, se pueden observar cilindros de glóbulos rojos en la orina, lo que es indicativo de que los glóbulos rojos están siendo filtrados de manera anormal a través de los glomérulos lesionados.
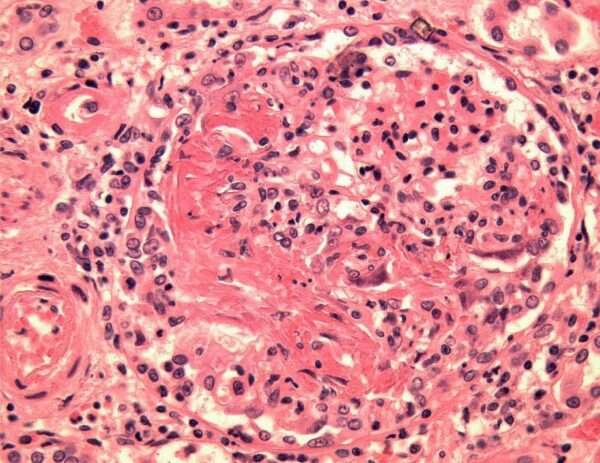
La lesión renal característica en esta enfermedad es una glomerulonefritis segmentaria necrotizante, que se refiere a la destrucción parcial de los glomérulos, con áreas de necrosis o muerte celular. Esta necrosis glomerular está a menudo acompañada de coagulación intravascular localizada, un fenómeno patológico donde se forman pequeños coágulos dentro de los vasos sanguíneos del riñón, lo que puede contribuir a la obstrucción y daño adicional del tejido renal. Cuando se realiza una biopsia renal en estos pacientes, se pueden observar trombos dentro de los glomérulos, lo que confirma el diagnóstico de una glomerulonefritis necrotizante asociada con esta vasculitis.
Diagnóstico diferencial
El diagnóstico diferencial de la granulomatosis eosinofílica con poliangeítis puede resultar complicado debido a la similitud con otras enfermedades vasculíticas y autoinmunitarias. Una de las principales dificultades es la distinción con la granulomatosis con poliangeítis, dado que ambas comparten características clínicas similares, como la afectación multisistémica y la presencia de vasculitis. Sin embargo, existen diferencias clave entre estas dos condiciones que permiten realizar una diferenciación precisa.
Una diferencia crítica es que la granulomatosis eosinofílica con poliangeítis no está asociada con la enfermedad crónica destructiva del tracto respiratorio superior que es una característica prominente de la granulomatosis con poliangeítis. En esta última, los pacientes suelen presentar síntomas como sinusitis crónica, otitis media y úlceras nasales, lo cual es menos común o ausente en la granulomatosis eosinofílica con poliangeítis. Además, otro aspecto fundamental para diferenciar estas dos enfermedades es la ausencia de inflamación granulomatosa en la granulomatosis eosinofílica con poliangeítis, mientras que en la granulomatosis con poliangeítis se observa la presencia de granulomas, que son agregados de células inflamatorias, en los tejidos afectados. Esta distinción histopatológica es clave, ya que los granulomas son un hallazgo característico que no se encuentra en la granulomatosis eosinofílica con poliangeítis.
Otro diagnóstico diferencial importante es la poliarteritis nodosa, ya que ambas enfermedades involucran la inflamación de los vasos sanguíneos. No obstante, la poliarteritis nodosa tiende a afectar principalmente las arterias medianas y grandes, y se caracteriza por la ausencia de los anticuerpos anticitoplasma de neutrófilos, que son fundamentales en la granulomatosis eosinofílica con poliangeítis. Además, los pacientes con poliarteritis nodosa no presentan las alteraciones típicas en los riñones, como la glomerulonefritis necrotizante que se observa en la granulomatosis eosinofílica con poliangeítis.
Otro trastorno poco frecuente que puede imitar la granulomatosis eosinofílica con poliangeítis es el síndrome de COPA, que es un trastorno autoinflamatorio multisistémico de origen genético. Este síndrome se caracteriza por hemorragias pulmonares recurrentes, un hallazgo que puede ser indistinguible de la granulomatosis eosinofílica con poliangeítis en algunos casos. El síndrome de COPA puede presentar autoanticuerpos en el suero, lo que complica aún más el diagnóstico diferencial. Sin embargo, a diferencia de la granulomatosis eosinofílica con poliangeítis, el síndrome de COPA tiene un inicio precoz, generalmente en la infancia, y suele presentarse con antecedentes familiares notables de la enfermedad. Este patrón hereditario y el curso clínico temprano en la vida pueden ayudar a diferenciarlo de la granulomatosis eosinofílica con poliangeítis, que generalmente afecta a adultos.
Tratamiento
El tratamiento de la granulomatosis eosinofílica con poliangeítis sigue en gran medida las mismas pautas que el tratamiento de la granulomatosis con poliangeítis, dado que ambas son enfermedades autoinmunitarias que involucran vasculitis y comparten características clínicas similares, como la afectación pulmonar y renal. En particular, los pacientes con formas graves de la enfermedad, que habitualmente incluyen hemorragias pulmonares y glomerulonefritis, requieren un tratamiento urgente para inducir la remisión de la enfermedad y controlar la inflamación sistémica.
El tratamiento inicial generalmente consiste en el uso de corticosteroides, que tienen un potente efecto antiinflamatorio e inmunosupresor. Los corticosteroides se utilizan para reducir rápidamente la inflamación y controlar los síntomas agudos de la enfermedad. Sin embargo, debido a la naturaleza agresiva de la granulomatosis eosinofílica con poliangeítis en su forma grave, los corticosteroides suelen ser acompañados por agentes inmunosupresores más específicos, como la ciclofosfamida o el rituximab.
La ciclofosfamida es un fármaco quimioterápico que suprime la actividad del sistema inmunológico al reducir la proliferación de linfocitos, las células que participan en el proceso inflamatorio de la vasculitis. Por otro lado, el rituximab es un anticuerpo monoclonal que se dirige contra los linfocitos B, que son responsables de la producción de los anticuerpos que desencadenan la inflamación en las vasculitis asociadas a ANCA. Ambos medicamentos son eficaces para inducir la remisión en los casos más graves de la enfermedad, como aquellos que presentan hemorragias pulmonares masivas o insuficiencia renal aguda debido a la glomerulonefritis.
Una vez alcanzada la remisión de la enfermedad, es necesario continuar con un tratamiento de mantenimiento para evitar recaídas. El tratamiento de mantenimiento tiene como objetivo mantener la respuesta terapéutica a largo plazo y minimizar los efectos secundarios de los tratamientos intensivos utilizados en la inducción de la remisión. El rituximab es considerado la opción de primera línea para el mantenimiento, ya que su capacidad para reducir los niveles de linfocitos B y, por lo tanto, suprimir la producción de anticuerpos patológicos, lo convierte en una herramienta eficaz en la prevención de recaídas.
Si el rituximab no está disponible o no es adecuado para un paciente específico, existen opciones de tratamiento de segunda línea, como la azatioprina, el metotrexato y el micofenolato de mofetilo. Estos fármacos también tienen propiedades inmunosupresoras y son capaces de mantener la remisión en los pacientes con granulomatosis eosinofílica con poliangeítis, aunque sus mecanismos de acción son diferentes al del rituximab. La azatioprina y el metotrexato inhiben la proliferación de células inmunitarias, mientras que el micofenolato actúa inhibiendo la síntesis de ácidos nucleicos en las células inmunes, lo que reduce la actividad del sistema inmunológico.
En los casos de vasculitis asociada a anticuerpos anticitoplasma de neutrófilos dirigida contra la mieloperoxidasa inducida por fármacos, el tratamiento debe centrarse en la interrupción inmediata del medicamento causante de la enfermedad. Si la vasculitis ha causado un daño significativo en órganos como los pulmones o los riñones, se requiere el uso de terapia inmunosupresora para prevenir un daño irreversible y controlar la inflamación sistémica.
Pronóstico
El pronóstico de la granulomatosis eosinofílica con poliangeítis depende en gran medida de un diagnóstico temprano y del tratamiento oportuno, ya que la intervención temprana puede mejorar significativamente los resultados en los pacientes. La importancia de un diagnóstico temprano radica en la capacidad de iniciar el tratamiento antes de que la enfermedad cause un daño irreversible en los órganos afectados, especialmente en los riñones y los pulmones.
En términos generales, los pacientes con granulomatosis eosinofílica con poliangeítis tienen un pronóstico más favorable cuando la enfermedad se diagnostica en etapas tempranas, ya que el tratamiento precoz puede controlar la inflamación y prevenir la progresión de la vasculitis. Sin embargo, cuando el diagnóstico se retrasa, los pacientes corren un mayor riesgo de desarrollar complicaciones graves, como insuficiencia renal crónica, daño pulmonar significativo o problemas cardíacos. Esto se debe a que la enfermedad tiende a ser más agresiva si no se trata adecuadamente desde el principio, lo que puede llevar a un daño irreversible en los órganos.
Una de las diferencias más importantes en el pronóstico de los pacientes con granulomatosis eosinofílica con poliangeítises la fibrosis renal. En los pacientes cuyo diagnóstico se retrasa, la biopsia renal muestra con mayor frecuencia una fibrosis significativa. La fibrosis es el resultado de un proceso de cicatrización en los riñones que ocurre cuando el tejido renal se daña de manera crónica. En el caso de la granulomatosis eosinofílica con poliangeítis, la inflamación crónica de los glomérulos, que son las unidades filtrantes del riñón, puede inducir la formación de tejido cicatricial. A medida que este proceso progresa, los riñones pierden su capacidad para filtrar la sangre de manera eficiente, lo que puede dar lugar a insuficiencia renal.
Por el contrario, en los pacientes con diagnóstico más temprano, se pueden tomar medidas para controlar la inflamación antes de que se produzca un daño renal significativo, lo que mejora las posibilidades de preservar la función renal a largo plazo. Si la enfermedad se detecta y se trata en sus primeras etapas, la fibrosis renal puede ser limitada o incluso prevenirse, lo que lleva a una mejor función renal y a un pronóstico más favorable.
Comparado con los pacientes que padecen granulomatosis con poliangeítis, aquellos que presentan granulomatosis eosinofílica con poliangeítis son más propensos a desarrollar fibrosis renal, debido principalmente a que la enfermedad en estos pacientes tiende a ser diagnosticada en fases más avanzadas. La granulomatosis con poliangeítis generalmente presenta síntomas más específicos y evidentes, como la afectación crónica del tracto respiratorio superior, lo que facilita su diagnóstico temprano en comparación con la granulomatosis eosinofílica con poliangeítis, que puede manifestarse de manera más insidiosa y con síntomas menos específicos, lo que retrasa la identificación de la enfermedad.

Fuente y lecturas recomendadas:
- Hellmich B et al. EULAR recommendations for the management of ANCA-associated vasculitis: 2022 update. Ann Rheum Dis. 2023:ard-2022-223764. [PMID: 36927642]
Aprende administración paso a paso

ADMINISTRACION DESDE CERO
Originally posted on 17 de marzo de 2025 @ 10:33 AM