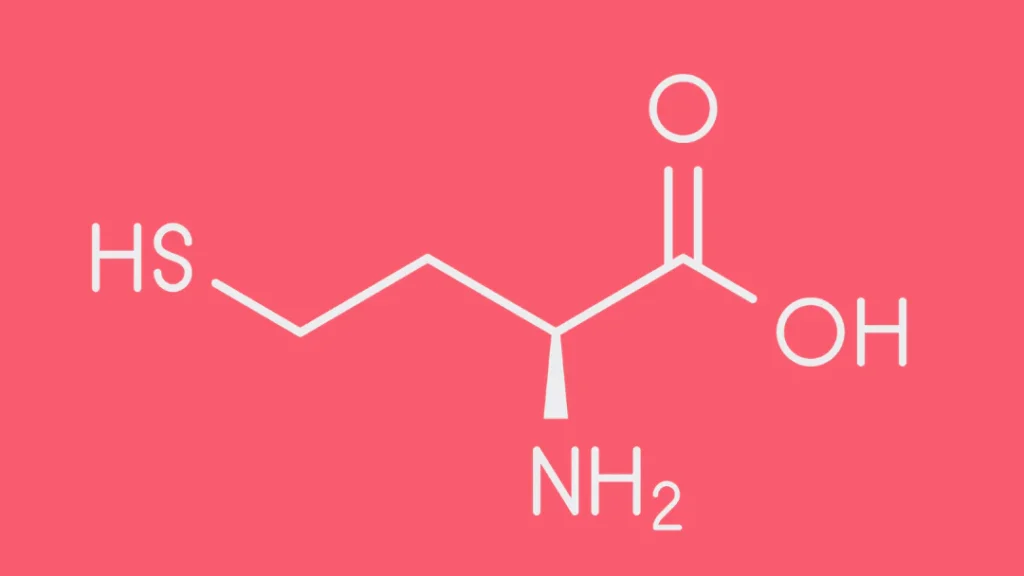
Los pacientes que presentan evidencia clínica y angiográfica de enfermedad arterial coronaria (EAC), junto con enfermedades cerebrovasculares, vasculares periféricas y tromboembolismo venoso (TEV), tienden a tener niveles más altos de homocisteína plasmática en comparación con aquellos que no padecen estas enfermedades vasculares. De igual manera, la elevación de la homocisteína en la sangre incrementa el riesgo de desarrollar demencia tipo Alzheimer en adultos mayores. Aunque inicialmente se pensaba que estos efectos podrían estar relacionados, al menos en parte, con los heterocigotos para la deficiencia de la cistationina beta-sintasa, existe poca evidencia que respalde esta hipótesis. En realidad, un factor crucial que conduce a la hiperhomocisteinemia es la deficiencia de ácido fólico.
Además, la piridoxina (vitamina B6) y la vitamina B12 desempeñan roles esenciales en el metabolismo de la metionina. La deficiencia de cualquiera de estas vitaminas puede provocar la acumulación de homocisteína en el organismo. Diversos genes influyen en la utilización de estas vitaminas y pueden predisponer a su deficiencia. Un ejemplo de ello es la presencia de uno, o incluso dos copias, de un alelo que causa termolabilidad de la metilenotetrahidrofolato reductasa, lo cual aumenta la predisposición a niveles elevados de homocisteína en ayunas.
Tanto las deficiencias nutricionales como la mayoría de las deficiencias genéticas de estas vitaminas pueden corregirse mediante la suplementación dietética con ácido fólico, y en caso de niveles bajos de suero, con vitaminas B6 y B12. Los cereales están fortificados con ácido fólico, lo cual ha contribuido a disminuir la prevalencia de esta deficiencia. Sin embargo, la terapia con vitaminas del grupo B y ácido fólico puede reducir de manera significativa los niveles de homocisteína, pero no parece disminuir el riesgo ni de tromboembolismo venoso ni de complicaciones asociadas con la enfermedad arterial coronaria.
La hiperhomocisteinemia también es frecuente en pacientes con enfermedad renal crónica terminal. En la población general, los niveles elevados de homocisteína se correlacionan con el deterioro cognitivo, lo que sugiere una relación entre el exceso de esta molécula y el riesgo de desarrollar trastornos neurológicos. En resumen, si bien la homocisteína elevada es un factor de riesgo asociado con diversas enfermedades vasculares y cognitivas, la corrección de sus niveles mediante suplementación vitamínica podría no ser suficiente para prevenir todas las complicaciones clínicas relacionadas.
Manifestaciones clínicas
Hiperhomocisteinemia
La hiperhomocisteinemia es un estado caracterizado por niveles elevados de homocisteína en plasma. A pesar de que continúa el debate sobre si una leve elevación de la homocisteína predispone al desarrollo de enfermedades arteriales oclusivas, existe un consenso sobre su relación con diversos trastornos vasculares. En adultos con hiperhomocisteinemia, especialmente aquellos que presentan deficiencias asociadas de vitaminas como el ácido fólico, la piridoxina (vitamina B6) o la vitamina B12, es común observar síntomas que reflejan estas carencias vitamínicas. Estas deficiencias pueden alterar procesos metabólicos esenciales, entre ellos la conversión de homocisteína en otros compuestos importantes, lo que contribuye a la acumulación de esta sustancia en el cuerpo. La homocisteína elevada está implicada en un mayor riesgo de sufrir enfermedades cardiovasculares, debido a su capacidad para dañar el endotelio vascular y promover procesos trombogénicos, favoreciendo la formación de coágulos en las arterias y venas. Sin embargo, los mecanismos exactos mediante los cuales los niveles elevados de homocisteína contribuyen al daño arterial no están completamente claros y continúan siendo un tema de investigación.
Homocistinuria
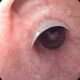
La homocistinuria, en su forma clásica, es un trastorno metabólico raro causado por la deficiencia de la cistationina beta-sintasa, una enzima clave en el ciclo de la metionina. Este trastorno sigue un patrón de herencia autosómica recesiva y tiene una prevalencia aproximada de 1 por cada 100,000 nacidos vivos. La consecuencia de esta deficiencia es la acumulación extrema de homocisteína tanto en plasma como en orina, lo que sirve como base para el diagnóstico de esta condición. A pesar de que la homocistinuria presenta algunas similitudes superficiales con el síndrome de Marfan, como la presencia de un tipo de cuerpo característico y la ectopia lentis (desplazamiento del cristalino), existen diferencias clínicas significativas. En la homocistinuria, es común que los pacientes experimenten deterioro cognitivo, una manifestación que rara vez se observa en el síndrome de Marfan. Además, los eventos cardiovasculares en los pacientes con homocistinuria se caracterizan por trombosis venosa y arterial recurrente, aunque la causa exacta de estas trombosis sigue siendo incierta.
El diagnóstico de homocistinuria debe ser considerado en pacientes jóvenes, particularmente aquellos en su segunda y tercera década de vida, que presenten trombosis arterial o venosa sin otros factores de riesgo obvios. En estos casos, la densidad mineral ósea también está reducida en los pacientes no tratados, lo que incrementa el riesgo de fracturas óseas. La esperanza de vida de los pacientes con homocistinuria es más corta, especialmente en aquellos que no responden al tratamiento con piridoxina (vitamina B6), con infartos de miocardio, accidentes cerebrovasculares y embolias pulmonares siendo las principales causas de mortalidad. El diagnóstico temprano de la homocistinuria se realiza a través del cribado neonatal para detectar niveles elevados de metionina, aunque los recién nacidos que responden a la piridoxina pueden no ser identificados en este proceso. Además, los individuos homocigotos para un alelo patogénico común, como el p.I278T, pueden presentar una notable variabilidad clínica, con algunos afectados sin manifestaciones clínicas evidentes en la adultez. Esto resalta la complejidad y la diversidad de la presentación clínica de la enfermedad, lo que complica su diagnóstico y manejo en diferentes grupos poblacionales.
Exámenes diagnósticos
A pesar de que se han identificado numerosas variaciones en el gen de la cistationina beta-sintasa (CBS), el análisis de los aminoácidos plasmáticos sigue siendo la prueba diagnóstica más adecuada para detectar trastornos relacionados con este gen. Esto se debe a que la medición directa de los niveles de homocisteína plasmática proporciona una indicación clara de la presencia de hiperhomocisteinemia, que es una manifestación clave de deficiencias en la CBS.
Para garantizar la precisión de los resultados, es fundamental que los pacientes sean estudiados después de haber suspendido la suplementación con ácido fólico o piridoxina durante al menos una semana. Esto se debe a que la suplementación vitamínica puede interferir con la medición, alterando los niveles de homocisteína en el plasma y dando lugar a resultados falsamente bajos. De este modo, se asegura que cualquier medición refleje un estado basal sin la influencia de factores externos.
Sin embargo, uno de los desafíos más grandes en la medición de homocisteína plasmática es que existen relativamente pocos laboratorios que ofrezcan ensayos altamente confiables para su cuantificación. La confiabilidad de estos ensayos depende de la correcta preparación y manejo de la muestra biológica. El proceso de separación del plasma de las células sanguíneas debe realizarse dentro de los primeros 30 minutos después de la extracción de la muestra. Esto es crucial porque, si las células sanguíneas permanecen en contacto con el plasma durante más tiempo, liberan homocisteína, lo que puede llevar a una elevación artificial de los niveles de la sustancia. Esta liberación de homocisteína por las células sanguíneas hace que los resultados obtenidos no sean representativos de los niveles verdaderos en el plasma, afectando así la precisión del diagnóstico.
Prevención
La prevención de la homocistinuria debido a la deficiencia de cistationina beta-sintasa se basa principalmente en el diagnóstico prenatal y, en algunos casos, la terminación de un embarazo afectado. Este enfoque preventivo es esencial, ya que la homocistinuria clásica, causada por la deficiencia de CBS, es un trastorno genético grave que, si no se detecta y trata de manera temprana, puede llevar a complicaciones severas, tales como trastornos cardiovasculares, problemas cognitivos y esqueléticos, así como un mayor riesgo de trombosis. Dado que la homocistinuria es una condición autosómica recesiva, el diagnóstico prenatal permite identificar a los fetos afectados antes del nacimiento, lo que brinda la opción de interrumpir el embarazo si así lo deciden los padres.
El diagnóstico prenatal generalmente se realiza mediante la identificación de los niveles elevados de metionina o homocisteína en el líquido amniótico o a través de la detección genética del alelo mutado en el feto. Sin embargo, la opción de terminar un embarazo afectado por esta condición es una decisión ética y personal que depende de los valores y creencias de los padres, así como del consejo genético que reciban. Este enfoque preventivo es actualmente la única manera de evitar la manifestación clínica de la homocistinuria CBS-deficiente en la descendencia.
Es fundamental que los profesionales de la salud estén bien informados sobre las leyes y regulaciones vigentes en su jurisdicción con respecto a la disponibilidad de anticoncepción de emergencia y la interrupción legal del embarazo. Estas leyes varían considerablemente entre diferentes países y regiones, por lo que es esencial que los clínicos comprendan las normativas locales y ofrezcan a los pacientes una orientación precisa sobre las opciones disponibles. En algunos lugares, las leyes sobre la interrupción del embarazo están restringidas a ciertos casos, como malformaciones fetales graves o riesgo para la vida de la madre, mientras que en otras, la legislación es más flexible. Además, el acceso a métodos de anticoncepción de emergencia es un componente clave para evitar embarazos no deseados, especialmente en situaciones de riesgo, y debería ser discutido abiertamente con los pacientes en edad reproductiva.
Tratamiento
Aproximadamente el 50% de los pacientes con homocistinuria debido a la deficiencia de cistationina beta-sintasa experimentan una mejora tanto clínica como bioquímica cuando reciben dosis farmacológicas de piridoxina (50–500 mg diarios por vía oral) y ácido fólico (5–10 mg diarios por vía oral). Este tratamiento específico es eficaz porque, en estos pacientes, la deficiencia de la enzima CBS responde positivamente a la administración de estas vitaminas, lo que permite una mejor metabolización de la homocisteína. Si el tratamiento se inicia en la infancia, es posible prevenir la aparición de daños neurológicos y otros problemas clínicos asociados, como los trastornos cardiovasculares y esqueléticos. La intervención temprana mediante el uso de piridoxina y ácido fólico permite normalizar los niveles de homocisteína en plasma, lo que detiene el progresivo deterioro que caracteriza a la enfermedad.
Sin embargo, existen pacientes que no responden a la piridoxina, es decir, que no presentan una mejora en los niveles de homocisteína ni en los síntomas clínicos tras el tratamiento con estas vitaminas. En estos casos, el manejo terapéutico se orienta a reducir la ingesta dietética de metionina, el aminoácido precursor de la homocisteína, al tiempo que se suplementa con cisteína, otro aminoácido necesario para equilibrar el metabolismo de la homocisteína. Este enfoque, que también debe comenzar en la infancia, es crucial para evitar la acumulación de homocisteína en el organismo y las complicaciones asociadas. La restricción de metionina y la suplementación con cisteína son esenciales para mantener niveles plasmáticos de homocisteína dentro de un rango seguro.
Adicionalmente, la vitamina betaina ha demostrado ser útil en pacientes con homocistinuria, ya que facilita una vía metabólica alternativa que permite reducir los niveles plasmáticos de metionina sin la necesidad de la enzima CBS defectuosa. La betaina actúa como un donador de metilo, lo que ayuda a la conversión de homocisteína en metionina, contribuyendo así a disminuir la acumulación de homocisteína y sus efectos perjudiciales. Esta estrategia es especialmente importante para aquellos pacientes cuya deficiencia de CBS no se resuelve con la piridoxina ni con la modificación dietética.
En cuanto al manejo de los pacientes con homocistinuria clásica que han experimentado trombosis venosa, estos pacientes requieren tratamiento anticoagulante para prevenir la formación de nuevos coágulos y tratar las complicaciones relacionadas con la trombosis. Sin embargo, es importante señalar que no existen estudios que respalden el uso profiláctico de medicamentos anticoagulantes como la warfarina o de agentes antiplaquetarios en estos pacientes, lo que indica que el tratamiento debe ser personalizado según las circunstancias clínicas de cada paciente. Esto resalta la necesidad de un enfoque cuidadoso y especializado en el manejo de la homocistinuria, considerando tanto la respuesta a las terapias específicas como los riesgos asociados con las complicaciones trombóticas.


Fuente y lecturas recomendadas:
- Bublil EM et al. Classical homocystinuria: from cystathionine beta-synthase deficiency to novel enzyme therapies. Biochimie. 2021;173:48. [PMID: 31857119]
- Marelli C et al; E-HOD Consortium. Clinical and molecular characterization of adult patients with late-onset MTHFR deficiency. J Inherit Metab Dis. 2021;44:777. [PMID: 33089527]
- Poddar R. Hyperhomocysteinemia is an emerging comorbidity in ischemic stroke. Exp Neurol. 2021;336:113541. [PMID: 33278453]
- Truitt C et al. Health functionalities of betaine in patients with homocystinuria. Front Nutr. 2021;8:690359. [PMID: 34568401]
- Weber Hoss GR et al. Classical homocystinuria: a common inborn error of metabolism? An epidemiological study based on genetic databases. Mol Genet Genomic Med. 2020;8:e1214. [PMID: 32232970]
Aprende administración paso a paso

ADMINISTRACION DESDE CERO