La esclerosis sistémica es un trastorno raro y crónico que se caracteriza por la fibrosis difusa de la piel y los órganos internos, lo que provoca una alteración significativa en la estructura y la función de estos tejidos. En esta enfermedad, el tejido conectivo de diversas partes del cuerpo experimenta un proceso anómalo de cicatrización, lo que lleva a la acumulación de colágeno en exceso, provocando un endurecimiento y un funcionamiento deficiente de los órganose afectados.
Esta patología suele manifestarse entre las tres y cinco décadas de vida, y afecta con mayor frecuencia a las mujeres, siendo estas dos o tres veces más propensas a desarrollar la enfermedad que los hombres. Aunque la esclerosis sistémica es relativamente rara, es una de las enfermedades autoinmunes más complejas debido a su capacidad para afectar de manera múltiple a diversos sistemas orgánicos, lo que complica su diagnóstico y tratamiento.
Existen dos formas principales de la enfermedad: la esclerosis sistémica limitada y la esclerosis sistémica difusa. Aproximadamente el 80% de los pacientes diagnosticados presentan la forma limitada, mientras que el 20% restante corresponde a la forma difusa. La principal distinción entre ambas formas radica en la extensión del involucramiento de la piel. En la esclerosis sistémica limitada, la fibrosis afecta principalmente a la piel de las manos, los brazos y la cara, mientras que en la forma difusa, el compromiso de la piel es más extenso e incluye áreas más grandes del cuerpo, lo que generalmente implica un curso más agresivo de la enfermedad.
Ambas formas de la esclerosis sistémica pueden implicar una afectación multisistémica de los órganos internos, como los pulmones, el corazón, los riñones y el tracto gastrointestinal. Sin embargo, las personas con esclerosis sistémica limitada generalmente tienen un mejor pronóstico que aquellas con la forma difusa. Esto se debe a que las complicaciones graves, como enfermedades pulmonares o renales que amenazan la vida, son menos comunes en la forma limitada. En cambio, en la esclerosis sistémica difusa, la progresión más rápida de la fibrosis y la implicación más extensa de los órganos internos hacen que las complicaciones graves sean más frecuentes.
Dentro de los síntomas que pueden desarrollarse en los pacientes con esclerosis sistémica limitada, la hipertensión pulmonar es particularmente característica. Esta afección, que involucra un aumento de la presión en las arterias pulmonares, es un factor de riesgo significativo para la insuficiencia cardíaca y otras complicaciones pulmonares. Por lo tanto, es fundamental que los pacientes con esclerosis sistémica limitada se sometan a una evaluación pulmonar regular para detectar y manejar adecuadamente cualquier afectación en los pulmones. Las pruebas de función pulmonar, la tomografía computarizada de alta resolución (HRCT) del tórax y la ecocardiografía son herramientas esenciales para evaluar la involucración cardiopulmonar, permitiendo así un diagnóstico temprano y la implementación de tratamientos apropiados.
Manifestaciones clínicas
Esclerosis sistémica limitada
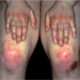
La esclerosis sistémica limitada es una variante de la esclerosis sistémica caracterizada por un compromiso cutáneo más restringido, que afecta principalmente la piel de la cara, el cuello y las áreas distales de los codos y las rodillas. A diferencia de la forma difusa, donde la fibrosis cutánea se extiende más ampliamente por todo el cuerpo, en la forma limitada la fibrosis es mucho más localizada. Sin embargo, a pesar de la menor extensión cutánea, la esclerosis sistémica limitada puede involucrar múltiples sistemas internos, y sus efectos pueden ser profundamente debilitantes, particularmente en lo que respecta a los vasos sanguíneos, el tracto gastrointestinal y los pulmones.
El fenómeno de Raynaud, que es la respuesta anormal de los vasos sanguíneos a los cambios en la temperatura o al estrés, es típicamente la manifestación inicial en la esclerosis sistémica limitada. En este fenómeno, los dedos de las manos y los pies se vuelven pálidos, fríos y pueden incluso adoptar un tono azulado como resultado de la vasoconstricción excesiva. Este síntoma puede preceder a otros signos clínicos de la enfermedad por varios años, lo que dificulta el diagnóstico temprano. Aunque el fenómeno de Raynaud puede ocurrir de forma aislada, en los pacientes con esclerosis sistémica limitada a menudo se observa en conjunto con otros síntomas que forman el síndrome CREST. El síndrome CREST es un conjunto de manifestaciones clínicas que incluyen calcinosis cutánea, fenómeno de Raynaud, trastornos de motilidad esofágica, esclerodactilia (endurecimiento de los dedos) y telangiectasias (dilatación de los vasos sanguíneos superficiales). Estos síntomas, aunque no necesariamente presentes todos en un mismo paciente, se asocian fuertemente con la forma limitada de la esclerosis sistémica.
A pesar de su naturaleza más localizada en términos de compromiso de la piel, los pacientes con esclerosis sistémica limitada tienen una mayor susceptibilidad a ciertas complicaciones graves en comparación con los pacientes que padecen la forma difusa. En particular, son más propensos a sufrir isquemia digital, lo que puede llevar a la pérdida de los dedos debido a la falta de suministro sanguíneo adecuado. Esta complicación puede resultar de la constricción crónica de los vasos sanguíneos en las extremidades, lo que interfiere con la circulación normal. Además, los pacientes con esclerosis sistémica limitada tienen un mayor riesgo de desarrollar hipertensión pulmonar, una afección en la que la presión en las arterias pulmonares se eleva de forma anormal, lo que puede llevar a insuficiencia cardíaca y otros problemas pulmonares graves.
La disfunción en el tracto gastrointestinal también es común en la esclerosis sistémica limitada. La hipomotilidad del intestino, que es una disminución en la capacidad del tracto gastrointestinal para mover los alimentos y los desechos, puede causar estreñimiento alternado con diarrea, malabsorción debido al crecimiento bacteriano excesivo, pseudo-obstrucción intestinal y distensión severa del intestino con posible ruptura. La dismotilidad esofágica es particularmente prominente, y los pacientes a menudo experimentan disfagia, que es la dificultad para tragar, así como síntomas de reflujo gastroesofágico debido a la alteración en los movimientos del esófago y, en etapas más avanzadas, por la fibrosis que se desarrolla en este órgano. La fibrosis en el tracto gastrointestinal conduce a una atrofia progresiva de los tejidos, lo que reduce aún más la motilidad y contribuye a los síntomas digestivos.
Además, la esclerosis sistémica limitada puede resultar en la formación de divertículos de gran tamaño en el intestino delgado (principalmente en el yeyuno y el íleon) y en el colon. Estos divertículos, que son pequeñas bolsas formadas en las paredes del intestino, pueden ser causados por la debilidad de las paredes del tracto gastrointestinal debido a la fibrosis y la atrofia muscular. Los divertículos pueden causar problemas adicionales, como infecciones o obstrucciones intestinales.
Esclerosis sistémica difusa
La esclerosis sistémica difusa es una variante de la enfermedad caracterizada por una progresión más rápida y extensa de la fibrosis, que afecta no solo la piel, sino también los órganos internos, lo que la distingue de la forma limitada, donde la afectación cutánea es más localizada. En la esclerosis sistémica difusa, los primeros síntomas suelen incluir poliartralgia (dolores en varias articulaciones), pérdida de peso y malestar general, manifestaciones que son comunes en esta forma de la enfermedad, pero mucho menos frecuentes en la forma limitada. Estos síntomas iniciales pueden ser indicativos de una respuesta inflamatoria generalizada y de un compromiso más amplio del sistema inmunológico, lo que señala una evolución más agresiva de la enfermedad.
A diferencia de la esclerosis sistémica limitada, que afecta principalmente la piel de las extremidades distales, la forma difusa de la enfermedad puede involucrar la piel del tronco y las extremidades proximales, lo que refleja una extensión más amplia del proceso fibrosante. Inicialmente, los cambios en la piel pueden incluir un edema subcutáneo no fóveo (es decir, que no deja una impresión al presionar), asociado con picazón, lo que indica la presencia de inflamación y acumulación de líquidos en los tejidos. Con el tiempo, este edema da paso a un endurecimiento de la piel, que se vuelve más gruesa y «pegada» a los planos subyacentes, perdiendo las pliegues normales de la piel. Este endurecimiento de la piel se debe al depósito excesivo de colágeno, una característica central de la esclerosis sistémica. Además, en la piel de los pacientes con esclerosis sistémica difusa es común observar telangiectasias (dilatación de los vasos sanguíneos superficiales), cambios en la pigmentación, tanto hiperpigmentación como hipopigmentación, y ulceraciones en los dedos de las manos, lo que puede contribuir a la disfunción de las extremidades. También es frecuente la presencia de calcificación subcutánea, un depósito de calcio en los tejidos, que es una complicación típica en esta forma de la enfermedad.
En algunos pacientes con esclerosis sistémica difusa, se pueden escuchar los llamados «frote tendinoso» sobre los antebrazos y las espinillas, una manifestación que ocurre cuando los tendones frotan contra la piel endurecida debido a la fibrosis. Sin embargo, no todos los pacientes presentan este signo.
Los órganos internos también están gravemente afectados en la esclerosis sistémica difusa. La fibrosis pulmonar y la enfermedad vascular pulmonar son comunes y se reflejan en una fisiología pulmonar restrictiva, es decir, una disminución de la capacidad de los pulmones para expandirse adecuadamente, lo que reduce la cantidad de oxígeno que se puede transferir a la sangre. Además, la capacidad de difusión pulmonar se ve reducida, lo que indica una afectación de las estructuras pulmonares responsables de intercambiar oxígeno y dióxido de carbono. En el corazón, se pueden observar varias anormalidades, como derrames pericárdicos (acumulación de líquido alrededor del corazón), bloqueo cardíaco, fibrosis miocárdica (cicatrización del músculo cardíaco) y insuficiencia cardíaca derecha secundaria a la hipertensión pulmonar, una complicación frecuente y grave que resulta de la elevación de la presión en los vasos pulmonares.
El tracto gastrointestinal también se ve afectado, de manera similar a la forma limitada de la enfermedad, con manifestaciones como dismotilidad esofágica, disfagia, reflujo gastroesofágico y alteraciones en la absorción de nutrientes, debido a la fibrosis y atrofia del músculo liso que controla los movimientos del intestino. Sin embargo, en la forma difusa, estos síntomas pueden ser más graves y estar más generalizados, afectando a todo el tracto gastrointestinal.
Una de las complicaciones más graves de la esclerosis sistémica difusa es la crisis renal esclerodérmica, una emergencia médica caracterizada por una proliferación intimal (engrosamiento de la capa interna) de las arterias renales pequeñas, lo que puede dar lugar a hipertensión arterial de aparición repentina y daño renal agudo. Esta condición es extremadamente grave y, si no se trata rápidamente, puede llevar a insuficiencia renal. Sin embargo, muchos casos de crisis renal pueden ser tratados de manera eficaz con inhibidores de la enzima convertidora de angiotensina, medicamentos que ayudan a controlar la hipertensión y prevenir el daño renal progresivo.
Exámenes diagnósticos
Los hallazgos de laboratorio en la esclerosis sistémica proporcionan información valiosa sobre el curso de la enfermedad y las posibles complicaciones. Uno de los hallazgos más comunes es una anemia leve, que a menudo está presente en los pacientes con esclerosis sistémica, debido a una disminución en la producción de glóbulos rojos o a un aumento en la destrucción de estos. La anemia leve puede estar relacionada con la inflamación crónica o con la afectación de los órganos internos, como los riñones o el tracto gastrointestinal, que pueden comprometer la absorción de nutrientes esenciales para la producción de glóbulos rojos.
En los casos de crisis renal, un hallazgo característico en el frotis de sangre periférica es la presencia de una anemia hemolítica microangiopática. Esta condición se produce debido al daño mecánico de los glóbulos rojos al pasar por pequeños vasos sanguíneos enfermos, que se encuentran alterados por el proceso fibrosante de la esclerosis sistémica. La hemólisis, es decir, la destrucción de los glóbulos rojos, es una complicación grave de la crisis renal, que puede llevar a una anemia más severa si no se trata de manera adecuada.
Un hallazgo de laboratorio importante en la esclerosis sistémica es la elevación de la velocidad de sedimentación globular, o ESR por sus siglas en inglés. Sin embargo, en la esclerosis sistémica, la elevación de la ESR es menos pronunciada que en otras enfermedades autoinmunes, lo que sugiere que el proceso inflamatorio puede ser menos activo o que la enfermedad tiene una naturaleza más fibrosante y crónica en lugar de una respuesta inflamatoria aguda.
La proteinuria leve, que generalmente es inferior a 2 gramos por 24 horas, es otro hallazgo frecuente en los análisis de orina. Esta cantidad de proteína en la orina indica que hay algún grado de daño renal, aunque generalmente no es tan grave como para indicar insuficiencia renal inminente. La proteinuria suele estar acompañada de pocos elementos celulares o cilindros en la orina, lo que sugiere que la afectación renal es leve y no hay signos de una inflamación aguda o de una infección renal significativa.
Desde el punto de vista inmunológico, los análisis serológicos revelan que los anticuerpos antinucleares (ANA) son casi siempre positivos en pacientes con esclerosis sistémica, y a menudo en altos títulos. Los anticuerpos antinucleares son un marcador clásico de enfermedades autoinmunes, y su presencia en la esclerosis sistémica indica que el sistema inmunológico está atacando de manera inapropiada los tejidos del cuerpo, en este caso, el colágeno y otros componentes del tejido conectivo.
Además de los ANA, otro marcador serológico importante es el anticuerpo contra la topoisomerasa III, conocido como anticuerpo anti-SCL-70. Este anticuerpo es encontrado en aproximadamente un tercio de los pacientes con esclerosis sistémica difusa y en un 20% de los pacientes con la forma limitada de la enfermedad. La presencia de anticuerpos anti-SCL-70 está asociada con un pronóstico más desfavorable, ya que sugiere una alta probabilidad de afectación grave de los órganos internos, especialmente los pulmones, con un riesgo elevado de desarrollar enfermedad pulmonar intersticial. Este hallazgo implica una forma de la enfermedad que puede progresar rápidamente y que presenta un mayor riesgo de complicaciones severas.
Por otro lado, los anticuerpos anticentromeros son comunes en aproximadamente el 50% de los pacientes con esclerosis sistémica limitada. Estos anticuerpos son altamente específicos de esta forma de la enfermedad, aunque también pueden encontrarse ocasionalmente en pacientes con síndromes de superposición, es decir, aquellos que presentan características de más de una enfermedad autoinmune. La presencia de anticuerpos anticentromeros se asocia típicamente con una forma menos grave de la enfermedad, con un mejor pronóstico, ya que la afectación de los órganos internos tiende a ser más limitada.
Un marcador adicional que tiene relevancia clínica en los pacientes con esclerosis sistémica es el anticuerpo contra la ARN polimerasa III. Este anticuerpo se presenta en el 10 al 20% de los pacientes con esclerosis sistémica y está vinculado a una forma de la enfermedad más agresiva, especialmente con un avance rápido de los cambios en la piel, crisis renal y un mayor riesgo de cánceres sólidos concomitantes, particularmente cáncer de mama. La presencia de anticuerpos anti-ARN polimerasa III indica un curso más rápido y potencialmente más grave de la enfermedad, con complicaciones renales agudas y un riesgo elevado de desarrollar ciertos tipos de cáncer, lo que hace que su detección sea importante para el manejo y seguimiento de estos pacientes.
Diagnóstico diferencial
En las etapas iniciales de su curso, la esclerosis sistémica puede causar confusión diagnóstica con otras afecciones que presentan el fenómeno de Raynaud, particularmente el lupus eritematoso sistémico, la enfermedad mixta del tejido conectivo y las miopatías inflamatorias. Estas enfermedades comparten algunos síntomas similares, como la vasoconstricción de los vasos sanguíneos en respuesta al frío o al estrés, lo que dificulta el diagnóstico temprano de la esclerosis sistémica. Además, en estas enfermedades autoinmunes, los pacientes pueden experimentar síntomas como fatiga, dolor articular y alteraciones cutáneas, lo que puede llevar a que el diagnóstico de esclerosis sistémica se pase por alto inicialmente.
El esclerodermia, una forma de esclerosis que a menudo se asocia con diabetes o infecciones crónicas, también puede confundirse con la esclerosis sistémica. Esta condición se caracteriza por un endurecimiento de la piel en áreas como la parte posterior del cuello, los hombros y la parte superior de la espalda, pero a diferencia de la esclerosis sistémica, no afecta a los dedos. Esta distinción es clave para diferenciarla de la esclerosis sistémica, que suele involucrar de manera prominente las extremidades. El esclerodermia asociado con la diabetes o infecciones no incluye las alteraciones internas típicas de la esclerosis sistémica, como la fibrosis pulmonar o la hipertensión pulmonar.
La fascitis eosinofílica, por su parte, es un trastorno raro que presenta endurecimiento de la piel, lo que puede parecerse a la esclerosis sistémica difusa. Sin embargo, en la fascitis eosinofílica, los cambios inflamatorios están limitados a la fascia (la capa de tejido conectivo que rodea los músculos y otros órganos), y no afectan la dermis y la epidermis, como ocurre en la esclerosis sistémica. Los pacientes con fascitis eosinofílica suelen presentar eosinofilia periférica (aumento de los eosinófilos en la sangre), una característica que no se encuentra en la esclerosis sistémica. Además, la fascitis eosinofílica no suele presentar el fenómeno de Raynaud, y los pacientes responden favorablemente al tratamiento con esteroides, lo que también ayuda a diferenciarla de la esclerosis sistémica. En algunos casos, la fascitis eosinofílica también se asocia con paraproteinemias, lo que no es típico de la esclerosis sistémica.
Otra afección que puede confundirse con la esclerosis sistémica es la escleromixedema, que se caracteriza por un engrosamiento de la piel difuso y un compromiso visceral. En este trastorno, la presencia de una paraproteína en la sangre (una proteína anómala producida por células plasmáticas) es un hallazgo distintivo, así como la ausencia del fenómeno de Raynaud. Además, la histología de la piel en la escleromixedema es diferente a la de la esclerosis sistémica, lo que ayuda a establecer el diagnóstico diferencial. Aunque la escleromixedema presenta síntomas cutáneos similares, la presencia de paraproteínas y la ausencia de otros hallazgos característicos de la esclerosis sistémica permiten una distinción diagnóstica.
La artropatía diabética o «cheiroartropatía» diabética es otra condición que puede simular la esclerodactilia, un síntoma característico de la esclerosis sistémica. La artropatía diabética suele desarrollarse en personas con diabetes mellitus de larga duración y mal controlada, especialmente cuando existe un daño crónico en las articulaciones de las manos. En esta condición, las articulaciones de los dedos se vuelven rígidas, lo que puede parecerse a la esclerodactilia, pero la diferencia clave radica en que la artropatía diabética está asociada con la diabetes, mientras que la esclerodactilia es una manifestación específica de la esclerosis sistémica.
La morfea y la esclerodermia lineal son trastornos que causan cambios esclerodérmicos limitados a áreas circunscritas de la piel, a diferencia de la esclerosis sistémica, que involucra una afectación generalizada de la piel y los órganos internos. La morfea se presenta típicamente como placas de piel endurecida y engrosada, generalmente en una sola área, y tiene un pronóstico mucho más favorable que la esclerosis sistémica. De manera similar, la esclerodermia lineal se presenta como bandas de piel endurecida, generalmente a lo largo de un solo lado del cuerpo, y también tiene un curso clínico mucho más benigno en comparación con la esclerosis sistémica.
Tratamiento
El tratamiento de la esclerosis sistémica, tanto en su forma difusa como limitada, se centra principalmente en los sistemas orgánicos involucrados, dado que, hasta la fecha, no existe una terapia efectiva que modifique el proceso subyacente de la enfermedad. La esclerosis sistémica es una enfermedad autoinmune crónica caracterizada por un proceso fibrosante que afecta la piel y varios órganos internos, y aunque no se dispone de un tratamiento que revierta esta fibrosis, las intervenciones terapéuticas dirigidas a manejar las manifestaciones específicas de los órganos afectados han mejorado considerablemente en los últimos años.
El tratamiento de la esclerosis sistémica difusa, dado que esta forma de la enfermedad tiene una progresión más rápida y un compromiso más generalizado de la piel y los órganos internos, los esfuerzos terapéuticos se orientan a controlar las complicaciones graves asociadas, tales como la fibrosis pulmonar, la hipertensión pulmonar y la insuficiencia renal. El uso de medicamentos inmunosupresores, como los antagonistas de la interleucina-6 y los inhibidores de la calcineurina, ha demostrado ser útil para reducir la inflamación y la progresión de la fibrosis en los pulmones y otros órganos internos. Asimismo, los tratamientos biológicos que modulan la respuesta inmune están siendo investigados como opciones terapéuticas, aunque los resultados son aún preliminares.
En la forma limitada de la esclerosis sistémica, los síntomas cutáneos suelen ser menos extensos, y la afectación de los órganos internos es más gradual. El tratamiento en estos casos también se enfoca en la gestión de las complicaciones, particularmente la hipertensión pulmonar, que es una de las principales preocupaciones para los pacientes con esclerosis sistémica limitada. Los vasodilatadores, como los inhibidores de la fosfodiesterasa-5 y los antagonistas de los receptores de endotelina, son eficaces para tratar la hipertensión pulmonar, y su uso ha mejorado significativamente la calidad de vida de los pacientes con esta complicación. Además, el manejo de la disfunción esofágica, los problemas gastrointestinales y las alteraciones en la motilidad también son aspectos importantes del tratamiento en la esclerosis sistémica limitada.
En general, no se ha demostrado que el uso de esteroides, como la prednisona, tenga un papel beneficioso en el tratamiento de la esclerosis sistémica, excepto en casos específicos. Los esteroides pueden ser útiles en pacientes con miopatía inflamatoria asociada a la enfermedad, ya que ayudan a reducir la inflamación y mejorar la fuerza muscular. Sin embargo, el uso de prednisona en dosis mayores a 15 mg diarios se ha relacionado con un aumento significativo del riesgo de desarrollar una crisis renal esclerodérmica, una complicación potencialmente fatal de la enfermedad, que se caracteriza por un daño renal agudo asociado a hipertensión severa.
-
Enfermedad cutánea: La afectación significativa de la piel, una característica prominente en muchas formas de esclerosis sistémica, puede beneficiarse de tratamientos inmunosupresores. Medicamentos como el metotrexato y el mofetil de micofenolato han demostrado ser útiles para controlar el engrosamiento cutáneo, especialmente si se inician lo más temprano posible en el curso de la enfermedad. El metotrexato, administrado por vía oral una vez por semana a dosis que se titulan hasta 15 o 20 mg, es una opción común, particularmente eficaz en pacientes con artritis asociada a la esclerosis sistémica. Por otro lado, el mofetil de micofenolato, cuya dosis diaria se puede titrar hasta los 2000 o 3000 mg, es preferido en aquellos pacientes que presentan enfermedad pulmonar intersticial, ya que tiene efectos protectores sobre los pulmones. En casos más graves y refractarios, las terapias como la inmunoglobulina intravenosa, rituximab y tocilizumab pueden ser opciones adicionales, especialmente en pacientes que no responden a los tratamientos estándar. La elección del tratamiento se adapta en función de las manifestaciones sistémicas asociadas, lo que subraya la importancia de un enfoque personalizado y multidisciplinario.
-
Crisis renal: La crisis renal esclerodérmica es una emergencia médica que requiere un tratamiento urgente y agresivo, particularmente cuando se asocia con una hipertensión grave. El tratamiento de la hipertensión en la crisis renal debe iniciarse en el entorno hospitalario, con medicamentos inhibidores de la enzima convertidora de angiotensina (ECA), como el captopril, que se administra por vía oral a dosis iniciales de 25 mg cada 6 horas. Esta dosis puede aumentarse gradualmente hasta un máximo de 100 mg cada 6 horas, según la tolerancia del paciente y la respuesta clínica. Los inhibidores de la ECA son esenciales para reducir la presión arterial y prevenir el daño renal adicional. La intervención temprana con este enfoque terapéutico ha demostrado ser crucial para mejorar los resultados en pacientes con crisis renal, evitando así la insuficiencia renal irreversible y la necesidad de tratamientos más invasivos, como la diálisis.
-
Enfermedad gastrointestinal: La afectación gastrointestinal, especialmente el reflujo esofágico y la hipomotilidad esofágica, es común en los pacientes con esclerosis sistémica. El reflujo esofágico puede ser controlado significativamente mediante el uso de inhibidores de la bomba de protones (IBP), como el omeprazol, que inhiben de manera casi completa la producción de ácido gástrico, reduciendo así la irritación del esófago y el riesgo de cicatrización. Los pacientes deben evitar las comidas nocturnas para disminuir el riesgo de reflujo. Además, el uso de IBP en dosis de 20 a 40 mg al día es efectivo para tratar la esofagitis refractaria asociada con esta enfermedad. Los pacientes con retraso en el vaciamiento gástrico pueden mejorar su nutrición y mantener un peso adecuado si siguen una dieta de comidas pequeñas y frecuentes, además de permanecer en posición vertical durante al menos dos horas después de comer. Para los casos de disfagia debida a hipomotilidad esofágica, los agentes procinéticos orales, como el metoclopramida (10 mg cuatro veces al día), pueden ser útiles para mejorar la motilidad esofágica y aliviar los síntomas. Si estos fármacos no son efectivos, la eritromicina (250 mg tres veces al día) puede considerarse como alternativa. En los casos de malabsorción causados por un sobrecrecimiento bacteriano en el intestino, los antibióticos como el rifaximina (550 mg tres veces al día) pueden ser prescritos de manera cíclica para tratar la infección bacteriana, lo que puede mejorar los síntomas de malabsorción y ayudar a restaurar una adecuada absorción de nutrientes.
-
Enfermedad pulmonar: La enfermedad pulmonar intersticial es una manifestación común y grave de la esclerosis sistémica, que puede provocar disnea (dificultad para respirar) y deterioro en las pruebas de función pulmonar. El tratamiento dirigido a esta complicación busca mejorar la función pulmonar y aliviar los síntomas respiratorios. El mofetil de micofenolato, administrado en dosis de 1000 a 1500 mg por vía oral dos veces al día, ha demostrado ser eficaz de manera modesta para mejorar la disnea y los resultados en las pruebas de función pulmonar en pacientes con enfermedad pulmonar intersticial asociada a la esclerosis sistémica. Este medicamento, al actuar como un inmunosupresor, ayuda a reducir la inflamación y la fibrosis en los pulmones, lo que puede mejorar la respiración y prevenir un mayor daño pulmonar.
Por otro lado, la ciclofosfamida, aunque también tiene eficacia en el tratamiento de la enfermedad pulmonar intersticial, está asociada con mayores riesgos y efectos secundarios, lo que requiere una supervisión cuidadosa por parte de médicos experimentados en su uso. La ciclofosfamida, que actúa como un agente inmunosupresor potente, puede ser útil en casos más graves o refractarios, pero su toxicidad, particularmente en los riñones y la médula ósea, limita su uso a situaciones donde los beneficios superan los riesgos.
En pacientes que no responden a los tratamientos con mofetil de micofenolato o ciclofosfamida, el nintedanib, un inhibidor de múltiples tirosina quinasas, ha mostrado una eficacia importante al ralentizar la progresión de la enfermedad pulmonar asociada a la esclerosis sistémica. Este medicamento está aprobado por la Administración de Alimentos y Medicamentos (FDA) para esta indicación y es una opción valiosa cuando los tratamientos tradicionales no son efectivos o no se toleran.
El tocilizumab, un inhibidor de la interleucina 6 (IL-6), también ha demostrado ser útil para ralentizar la disminución de la función pulmonar en pacientes con esclerosis sistémica. La administración subcutánea de tocilizumab, a razón de 162 mg una vez por semana, puede ser una alternativa adecuada para aquellos pacientes que no toleran el mofetil de micofenolato, ofreciendo una opción de tratamiento adicional en casos de enfermedad pulmonar intersticial progresiva. Además, otros medicamentos inmunosupresores como la azatioprina y rituximab pueden ser considerados en el manejo de la enfermedad pulmonar asociada a la esclerosis sistémica, aunque su uso debe evaluarse cuidadosamente debido a los efectos secundarios potenciales y la respuesta clínica variable entre los pacientes.
En los casos más graves de enfermedad pulmonar, cuando la función pulmonar se deteriora significativamente, el trasplante pulmonar se considera una opción terapéutica. Los resultados de los trasplantes pulmonares en pacientes con esclerosis sistémica suelen ser favorables, y este procedimiento puede ofrecer una mejora considerable en la calidad de vida y en la supervivencia, especialmente en aquellos con enfermedad pulmonar intersticial avanzada que no responde a los tratamientos médicos.
-
Hipertensión pulmonar: La hipertensión pulmonar es otra complicación grave de la esclerosis sistémica que afecta principalmente a los vasos sanguíneos de los pulmones, causando un aumento de la presión en la arteria pulmonar y llevando a una disminución de la capacidad pulmonar y la insuficiencia del lado derecho del corazón. El tratamiento de la hipertensión pulmonar en la esclerosis sistémica se basa en gran medida en terapias que han demostrado eficacia en estudios de hipertensión pulmonar en general, independientemente de la causa subyacente.
Las opciones terapéuticas para la hipertensión pulmonar incluyen bloqueadores de los canales de calcio, como el amlodipino, nifedipino y el diltiazem de liberación prolongada, que actúan relajando los vasos sanguíneos y reduciendo la presión en los pulmones. Los inhibidores de la fosfodiesterasa tipo 5, como el sildenafil y el tadalafil, también se utilizan comúnmente para tratar la hipertensión pulmonar, ya que ayudan a dilatar los vasos sanguíneos pulmonares, mejorando el flujo sanguíneo y reduciendo la presión en las arterias pulmonares.
El riociguat, un estimulante de la guanilato ciclasa, y los antagonistas de los receptores de endotelina, como el bosentán, ambrisentán y macitentan, también son opciones terapéuticas para reducir la presión pulmonar y mejorar los síntomas en pacientes con hipertensión pulmonar asociada a la esclerosis sistémica. Estos medicamentos ayudan a reducir la vasoconstricción pulmonar y a mejorar la función del ventrículo derecho del corazón, que puede verse afectado en esta condición.
En casos de hipertensión pulmonar refractaria, es decir, cuando los tratamientos anteriores no han logrado controlar adecuadamente la presión pulmonar, los agonistas de la vía de la prostaciclina, como el epoprostenol, treprostinil, iloprost y selexipag, pueden ser necesarios. Estos medicamentos son potentes vasodilatadores que actúan en la vasculatura pulmonar para reducir la resistencia vascular pulmonar y mejorar la función respiratoria en pacientes con hipertensión pulmonar severa.
En los casos graves de esclerosis sistémica difusa que no responden a las terapias convencionales previamente mencionadas, como los inmunosupresores, el tratamiento con ablación mieloide seguido de un trasplante autólogo de células madre se ha considerado como una opción terapéutica superior en términos de eficacia. Este enfoque, aunque más invasivo y con un mayor potencial de toxicidad, ofrece ventajas en comparación con la inmunosupresión con ciclofosfamida, especialmente en pacientes con enfermedad avanzada y refractaria.
La esclerosis sistémica difusa, en su forma más severa, puede implicar una progresión rápida y debilitante de la enfermedad que afecta múltiples sistemas, como la piel, los pulmones, el corazón y los riñones. En estos casos, los tratamientos estándar con medicamentos inmunosupresores, como el mofetil de micofenolato o la ciclofosfamida, aunque efectivos en muchos pacientes, no siempre logran controlar adecuadamente la progresión de la enfermedad o la fibrosis en órganos clave. La ablación mieloide seguida de un trasplante autólogo de células madre es una estrategia que ha mostrado ser más eficaz para tratar a los pacientes con formas graves y resistentes de esclerosis sistémica, al abordar de manera más agresiva la disfunción inmunitaria subyacente.
La ablación mieloide implica la destrucción de las células madre hematopoyéticas del paciente a través de un régimen de quimioterapia intensa. Esto elimina el sistema inmunológico existente, que está desempeñando un papel crucial en el daño continuo de los tejidos. Posteriormente, se transfieren células madre hematopoyéticas autólogas, es decir, células madre obtenidas del propio paciente antes de la ablación. Estas células madre son capaces de regenerar un sistema inmunológico funcional que, en muchos casos, tiene una menor tendencia a atacar los tejidos sanos del cuerpo, lo que puede reducir significativamente la inflamación y la fibrosis asociadas con la esclerosis sistémica. Este enfoque tiene como objetivo «reiniciar» el sistema inmunológico del paciente, proporcionando una oportunidad para frenar la progresión de la enfermedad y mejorar la función de los órganos afectados.
Este tratamiento ha demostrado ser superior a la inmunosupresión convencional con ciclofosfamida en cuanto a la remisión o estabilización a largo plazo de la enfermedad. Los estudios han mostrado que, después de un trasplante autólogo de células madre, muchos pacientes experimentan una mejora sustancial en los síntomas y en las pruebas de función pulmonar, renal y cardiovascular, con una reducción de la fibrosis en los órganos afectados. A pesar de estos beneficios, el procedimiento conlleva un mayor riesgo de complicaciones, como infecciones graves, hemorragias y efectos secundarios derivados de la quimioterapia utilizada en la ablación mieloide.
La mayor toxicidad del trasplante autólogo de células madre en comparación con la inmunosupresión convencional se debe a la naturaleza intensiva del régimen de quimioterapia, que elimina temporalmente las células sanguíneas del paciente, lo que aumenta la vulnerabilidad a infecciones y otras complicaciones asociadas con la inmunodepresión. Además, el proceso de trasplante puede requerir un largo período de hospitalización y seguimiento cercano para monitorear la reconstitución del sistema inmunológico y detectar cualquier signo de rechazo o complicación.
Sin embargo, para los pacientes con esclerosis sistémica difusa grave y refractaria, que no han mostrado respuesta a los tratamientos convencionales y cuya calidad de vida y pronóstico están gravemente comprometidos, el trasplante de células madre autólogas puede ofrecer una oportunidad única para controlar la enfermedad de manera más eficaz.
Pronóstico
El pronóstico de la esclerosis sistémica varía considerablemente dependiendo de la gravedad de la enfermedad y de los órganos involucrados. En términos generales, la tasa de supervivencia a los diez años para los pacientes con esclerosis sistémica es de aproximadamente el 70%, lo que indica que, aunque esta enfermedad puede ser crónica y progresiva, muchos pacientes pueden sobrevivir y manejar su condición durante varios años con el tratamiento adecuado. No obstante, la evolución de la enfermedad está profundamente influenciada por la afectación de órganos internos, especialmente los pulmones, y la presencia de complicaciones graves.
Las enfermedades pulmonares son las principales causas de mortalidad en los pacientes con esclerosis sistémica. La fibrosis pulmonar, que es el endurecimiento y cicatrización del tejido pulmonar, y la hipertensión arterial pulmonar, que implica un aumento de la presión en las arterias pulmonares, son las complicaciones más comunes y peligrosas en estos pacientes. La fibrosis pulmonar puede dificultar la respiración, mientras que la hipertensión pulmonar sobrecarga el lado derecho del corazón, lo que puede llevar a insuficiencia cardiaca y, eventualmente, a la muerte si no se maneja adecuadamente. Estas complicaciones pulmonares son responsables de una parte significativa de las muertes asociadas con la esclerosis sistémica, ya que las alteraciones en la función pulmonar y cardiovascular pueden progresar rápidamente en algunos casos.
El pronóstico de los pacientes con esclerosis sistémica mejora considerablemente si la enfermedad no afecta gravemente a los órganos internos durante los primeros tres años tras el inicio de los síntomas. Esta ventana temporal de tres años es crucial porque en la mayoría de los casos, si no hay un daño significativo a los órganos principales, como los pulmones, los riñones y el corazón, durante este período inicial, los pacientes tienden a tener una evolución más favorable a largo plazo. Por el contrario, aquellos que desarrollan complicaciones graves dentro de los primeros años de la enfermedad tienen un pronóstico menos favorable, debido a la progresión de los daños orgánicos que afectan la supervivencia.
Además, se han realizado estudios pequeños que investigan el vínculo entre la esclerosis sistémica y la aparición simultánea de cáncer. En estos estudios, se ha observado que en algunos pacientes la esclerosis sistémica podría haberse desarrollado como una respuesta inmune dirigida hacia las células cancerosas. Este fenómeno sugiere que en ciertos casos, el sistema inmunológico podría haber activado una respuesta inflamatoria generalizada como mecanismo de defensa contra el cáncer, lo que podría contribuir al desarrollo de la enfermedad. Esta hipótesis es interesante, ya que implica que la esclerosis sistémica en algunos pacientes podría tener una etiología relacionada con el cáncer, lo que podría influir en el curso y la respuesta al tratamiento de la enfermedad.

Fuente y lecturas recomendadas:
- Pope JE et al. State-of-the-art evidence in the treatment of systemic sclerosis. Nat Rev Rheumatol. 2023;19:212. [PMID: 36849541]
- Volkmann ER et al. Systemic sclerosis. Lancet. 2023;401:304. [PMID: 36442487]
Aprende administración paso a paso

ADMINISTRACION DESDE CERO
Originally posted on 15 de marzo de 2025 @ 9:13 AM