El síndrome de Apert, también conocido como acrocefalosindactilia tipo 1, es una enfermedad genética rara que afecta el desarrollo del cráneo y la cara, así como también las extremidades. La causa subyacente del síndrome de Apert es una mutación en el gen FGFR2, que codifica un receptor de factor de crecimiento similar a la insulina.
El síndrome de Apert fue descrito por primera vez por el médico francés Eugène Apert en 1906. Apert era un médico pediatra y neurólogo que trabajaba en el Hospital Sainte-Eugénie en París. En su descripción original, Apert describió a dos pacientes con deformidades craneofaciales similares y reconoció la naturaleza hereditaria de la enfermedad. Aunque el síndrome lleva el nombre de Apert, no fue hasta la década de 1960 que se identificó la causa genética subyacente del síndrome. Hoy en día, el síndrome de Apert es ampliamente reconocido como una enfermedad genética rara causada por mutaciones en el gen FGFR2.
En condiciones normales, el FGFR2 se encarga de regular el crecimiento y la división celular durante el desarrollo fetal. Sin embargo, en el síndrome de Apert, la mutación en este gen provoca una activación excesiva de la señalización de FGFR2, lo que conduce a una fusión prematura de los huesos del cráneo y la cara, así como a la fusión de los dedos de las manos y los pies.
El síndrome de Apert se hereda de forma autosómica dominante, lo que significa que una sola copia mutada del gen FGFR2 es suficiente para causar la enfermedad. La mayoría de los casos son esporádicos, lo que significa que la mutación ocurre de forma aleatoria durante la formación del óvulo o del espermatozoide, y no se hereda de los padres. Sin embargo, algunas personas con síndrome de Apert tienen un padre afectado que también tiene la enfermedad.
Manifestaciones clínicas
Los signos y síntomas del síndrome de Apert pueden variar en gravedad y presentación, pero los más comunes incluyen:
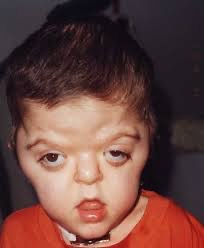
- Cabeza y cara: craneosinostosis (fusión prematura de los huesos del cráneo), hipertelorismo ocular (ojos muy separados), mandíbula pequeña y abombada, nariz ancha y prominente, labio y paladar hendido, y dientes apiñados.
- Extremidades: sindactilia (fusión de los dedos de las manos y/o los pies), manos y pies pequeños y anchos, dedos cortos y en forma de garra, y codos y rodillas doblados hacia afuera.
- Otras características: retraso en el desarrollo cognitivo, dificultad para hablar, problemas respiratorios y de audición, y riesgo aumentado de infecciones del oído y problemas dentales.
Exámenes complementarios
El diagnóstico del síndrome de Apert se basa en la observación de los signos y síntomas característicos, así como en pruebas genéticas para confirmar la mutación en el gen FGFR2. El tratamiento del síndrome de Apert es sintomático y puede incluir cirugía reconstructiva para corregir las anormalidades craneofaciales y de las extremidades, así como intervenciones para tratar otros problemas de salud asociados con la enfermedad.
Tratamiento
El tratamiento del síndrome de Apert se aborda en dos áreas principales: el tratamiento de las anomalías craneofaciales y el tratamiento de las anomalías de las extremidades.
Tratamiento de las anomalías craneofaciales:
La fusión prematura de los huesos del cráneo y la cara es la característica más distintiva del síndrome de Apert. La craneosinostosis puede provocar una presión intracraneal aumentada, lo que puede llevar a una variedad de problemas de salud graves, como hidrocefalia, problemas visuales y disminución de la función cerebral. El tratamiento de la craneosinostosis puede incluir lo siguiente:
- Cirugía craneofacial: La cirugía craneofacial se utiliza para separar los huesos fusionados y remodelar el cráneo y la cara. La cirugía temprana es fundamental para minimizar el riesgo de daño cerebral y visual y para permitir un mejor crecimiento del cerebro. La cirugía también puede ayudar a corregir otras anomalías faciales, como la hipertelorismo ocular, la mandíbula pequeña y abombada, y el labio y paladar hendido.
- Terapia ocular: En algunos casos, el síndrome de Apert puede afectar la función ocular y provocar problemas de visión, como estrabismo (ojos desalineados) o ambliopía (ojo perezoso). La terapia ocular, como los ejercicios oculares y los lentes correctivos, puede ser necesaria para corregir estos problemas.
Tratamiento de las anomalías de las extremidades:
Las personas con síndrome de Apert también pueden tener anomalías en las extremidades, como la sindactilia (fusión de los dedos de las manos y/o los pies), manos y pies pequeños y anchos, dedos cortos y en forma de garra, y codos y rodillas doblados hacia afuera. El tratamiento de las anomalías de las extremidades puede incluir:
- Cirugía de separación de los dedos: La cirugía de separación de los dedos se utiliza para dividir los dedos fusionados y crear dedos individuales. La cirugía se realiza típicamente en varias etapas, con una cirugía de seguimiento para remodelar los dedos y mejorar su apariencia y función.
- Cirugía de extensión de los dedos: La cirugía de extensión de los dedos se utiliza para alargar los dedos cortos y en forma de garra. Durante la cirugía, se insertan varillas metálicas en los huesos de los dedos y se estiran gradualmente para aumentar su longitud.
- Terapia ocupacional: La terapia ocupacional se utiliza para ayudar a mejorar la función de las manos y los dedos, y puede incluir ejercicios y técnicas de movimiento.
Otras intervenciones:
Las personas con síndrome de Apert también pueden requerir otras intervenciones para tratar problemas de salud asociados con la enfermedad, Estas pueden incluir:
- Tratamiento de la apnea del sueño: La apnea del sueño es un problema común en personas con síndrome de Apert debido a las anomalías en las vías respiratorias superiores. Los tratamientos pueden incluir el uso de una máscara CPAP (presión positiva continua en las vías respiratorias) durante el sueño para ayudar a mantener las vías respiratorias abiertas.
- Tratamiento de la hidrocefalia: La hidrocefalia es un problema común en personas con síndrome de Apert debido a la presión intracraneal aumentada. El tratamiento puede incluir la colocación de un shunt cerebral para drenar el exceso de líquido cerebral hacia el abdomen.
- Tratamiento dental: Las personas con síndrome de Apert pueden tener problemas dentales, como maloclusión, apiñamiento dental y retraso en la erupción dental. El tratamiento dental puede incluir la ortodoncia, la extracción dental y la reconstrucción dental.
Es importante destacar que el tratamiento del síndrome de Apert es un proceso a largo plazo y puede requerir varias intervenciones médicas y quirúrgicas a lo largo de la vida de la persona. Además, cada persona con síndrome de Apert es única y puede requerir un plan de tratamiento personalizado para abordar sus necesidades individuales.
En general, el pronóstico para las personas con síndrome de Apert depende de la gravedad de los síntomas y la efectividad del tratamiento. Con una atención médica adecuada, muchas personas con síndrome de Apert pueden llevar vidas saludables y productivas.

Síguenos en X: @el_homomedicus y @enarm_intensivo Síguenos en instagram: homomedicus y en Treads.net como: Homomedicus
Aprende administración paso a paso

ADMINISTRACION DESDE CERO