
La leucemia aguda es un tipo de malignidad que afecta a las células progenitoras hematopoyéticas, es decir, aquellas células precursoras de los diferentes tipos de células sanguíneas que se generan en la médula ósea. En esta enfermedad, las células inmaduras y malignas se multiplican de manera descontrolada, lo que provoca una sustitución de los elementos normales de la médula ósea. Esta proliferación anómala impide la producción adecuada de células sanguíneas normales, como glóbulos rojos, glóbulos blancos y plaquetas, lo que lleva a una serie de complicaciones hematológicas.
La mayoría de los casos de leucemia aguda surgen sin una causa clara y directa, es decir, se presentan de forma esporádica. Sin embargo, existen ciertos factores de riesgo que están asociados con un mayor riesgo de desarrollar esta enfermedad. La exposición a la radiación, por ejemplo, es uno de los factores que se ha identificado como leucomogénico, es decir, capaz de inducir la formación de leucemia. La radiación puede dañar el material genético de las células hematopoyéticas, lo que puede resultar en mutaciones y en una proliferación anómala de estas células.
Otro factor importante es la exposición a ciertas sustancias tóxicas, como el benceno. Este compuesto químico, que se encuentra en varios productos industriales y de consumo, también se ha relacionado con un aumento del riesgo de leucemia. El benceno, al ser un carcinógeno, puede alterar el ADN de las células hematopoyéticas, provocando transformaciones malignas.
Además, algunos tratamientos quimioterapéuticos, que son utilizados para tratar otros tipos de cáncer, también pueden inducir leucemia en un porcentaje significativo de pacientes. Entre estos fármacos se incluyen el ciclofosfamida, el melphalan, otros agentes alquilantes y el etopósido. Estos medicamentos, aunque efectivos para destruir células cancerosas, también pueden causar mutaciones en las células hematopoyéticas normales, lo que favorece el desarrollo de leucemias secundarias. Los casos de leucemia que ocurren después de la exposición a toxinas o quimioterapia suelen desarrollarse a partir de un cuadro precursor conocido como síndrome mielodisplásico, que se caracteriza por una producción anormal de células sanguíneas y una mayor susceptibilidad a la transformación leucémica.
En términos genéticos, las leucemias que se desarrollan tras la exposición a estos factores suelen presentar anomalías cromosómicas específicas. Las más comunes son las alteraciones en los cromosomas 5 y 7, que se encuentran frecuentemente en los pacientes con leucemia secundaria inducida por toxinas o quimioterapia. Estas anomalías cromosómicas afectan genes clave que regulan el ciclo celular y la apoptosis, favoreciendo la proliferación de células anormales. Por otro lado, en las leucemias asociadas con el uso de etopósido o antraciclinas, una característica genómica común es la presencia de alteraciones en el cromosoma 11q23, en particular en la región que alberga el locus MLL (mixed-lineage leukemia). Esta alteración génica se asocia con una reordenación de genes que contribuye al desarrollo de la leucemia al alterar los procesos de diferenciación celular.
La mayoría de los hallazgos clínicos en la leucemia aguda se deben a la sustitución de los elementos normales de la médula ósea por células malignas. Esta sustitución interfiere con la producción normal de las células sanguíneas, lo que lleva a una serie de manifestaciones clínicas, entre las que se incluyen anemia, neutropenia y trombocitopatía. Esto resulta en síntomas como fatiga, infecciones recurrentes y hemorragias. Menos comúnmente, las manifestaciones pueden involucrar la infiltración de órganos fuera de la médula ósea, como la piel, el tracto gastrointestinal o las meninges, lo que provoca una gama más amplia de síntomas dependiendo del órgano afectado.
La leucemia aguda tiene el potencial de ser curada, especialmente con la administración de quimioterapia combinada. El tratamiento efectivo depende de la rápida erradicación de las células leucémicas y de la restauración de la función hematopoyética normal. Sin embargo, el pronóstico varía según el subtipo de leucemia, la edad del paciente, las características genéticas y la respuesta al tratamiento.
El subtipo mieloblástico de la leucemia aguda, conocido como leucemia mieloide aguda (AML, por sus siglas en inglés), es predominantemente una enfermedad de adultos, con una mediana de edad en el momento del diagnóstico de 60 años. La incidencia de la AML aumenta con la edad, y su diagnóstico es más común en personas de mayor edad. En este subtipo, las células mieloides inmaduras se acumulan en la médula ósea, interrumpiendo la producción de células sanguíneas normales.
Por otro lado, la leucemia promielocítica aguda (APL) es una forma específica de la AML que se caracteriza por una translocación cromosómica t(15;17), que da lugar a la formación del gen de fusión PML-RAR-alfa. Este gen de fusión produce un bloqueo en la diferenciación de los precursores mieloides, lo que lleva a una acumulación de promielocitos en la médula ósea. Sin embargo, este bloqueo en la diferenciación puede ser superado con dosis farmacológicas de ácido retinoico, un tratamiento que ha revolucionado el pronóstico de los pacientes con APL, convirtiéndola en una de las leucemias más curables cuando se maneja adecuadamente.
En cuanto al subtipo linfoblástico, la leucemia linfoblástica aguda (ALL, por sus siglas en inglés) representa aproximadamente el 80% de los casos de leucemia aguda en niños. La incidencia máxima de ALL ocurre entre los 3 y los 7 años de edad, lo que la convierte en la leucemia más común en la infancia. Aunque la ALL es más prevalente en los niños, también se observa en adultos, donde constituye alrededor del 20% de los casos de leucemia aguda. En este subtipo, las células linfoides inmaduras (linfoblastos) invaden la médula ósea, lo que interfiere con la producción normal de células sanguíneas. El tratamiento de la ALL suele ser intensivo, y aunque es altamente curable en los niños, el pronóstico en los adultos tiende a ser más reservado.
Clasificación de las leucemias
La clasificación de las leucemias se basa en las características morfológicas, genéticas y biológicas de las células malignas involucradas, así como en el linaje celular afectado (mieloide o linfoide).
- Leucemia Mieloide Aguda (AML): Es un tipo de leucemia en la que las células mieloides inmaduras (precursores de glóbulos rojos, glóbulos blancos y plaquetas) se multiplican de forma descontrolada en la médula ósea. La AML afecta principalmente a adultos y tiene diversas subcategorías basadas en características morfológicas y genéticas.
- Leucemia Promielocítica Aguda (APL): Es una forma específica de AML caracterizada por una translocación cromosómica t(15;17), que produce el gen de fusión PML-RAR-alfa. Esta alteración genética interrumpe la diferenciación normal de los promielocitos, pero se puede tratar eficazmente con ácido retinoico, lo que ha mejorado significativamente el pronóstico.
- Leucemia Linfoblástica Aguda (ALL): Afecta principalmente a las células linfoides inmaduras (linfoblastos), predominando en niños, aunque también se observa en adultos. Se caracteriza por una rápida proliferación de linfoblastos en la médula ósea, que impide la producción de células sanguíneas normales. Su tratamiento suele ser intensivo y tiene una alta tasa de curación en niños.
- Leucemia Aguda de Línea Ambigua (ALAL): Es un subtipo raro de leucemia en la que las células malignas comparten características tanto mieloides como linfoides, lo que dificulta su clasificación en una de las categorías tradicionales. Se diagnostica cuando las células leucémicas no cumplen con los criterios claros para ser clasificadas como mieloides o linfoblásticas, y puede requerir estudios adicionales para su caracterización precisa.
Leucemia mieloide aguda
La leucemia mieloide aguda (LMA) es un tipo de leucemia caracterizado por la proliferación descontrolada de células mieloides inmaduras, lo que interfiere con la producción de células sanguíneas normales en la médula ósea. La clasificación de la LMA se basa principalmente en la identificación de anomalías cromosómicas y moleculares recurrentes que permiten agrupar los casos según su perfil genético y su pronóstico. Estas anomalías se pueden detectar mediante técnicas cito genéticas como el cariotipaje tradicional o la hibridación in situ con fluorescencia (FISH), que permiten observar cambios estructurales en los cromosomas. Además, las alteraciones moleculares se identifican mediante secuenciación dirigida o de todo el genoma del ADN tumoral.
En cuanto a los aspectos cito genéticos, las anomalías favorables incluyen translocaciones cromosómicas específicas, como la translocación t(8;21), que da lugar a la formación de una proteína quimérica RUNX1/RUNX1T1, y la inversión inv(16)(p13;q22), las cuales se observan en aproximadamente el 15% de los casos. Estas alteraciones se conocen como leucemias del «factor de unión al núcleo», ya que afectan genes clave en la regulación de la diferenciación celular. Los pacientes con estas anomalías tienen una probabilidad significativamente mayor de lograr un control tanto a corto como a largo plazo de la enfermedad, lo que les otorga un pronóstico favorable.
En contraste, las anomalías cito genéticas desfavorables están asociadas con un pronóstico muy pobre y representan aproximadamente el 25% de los casos de LMA. Estas incluyen translocaciones cromosómicas complejas como t(6;9), t(3;3) o inv(3), así como la translocación t(v;11q23), que afecta el locus MLL (mixed-lineage leukemia). También se incluyen en este grupo monosomías aisladas de los cromosomas 5 o 7, la presencia de dos o más monosomías adicionales, o tres o más anomalías cito genéticas separadas. Estas alteraciones cromosómicas están asociadas con un comportamiento agresivo de la enfermedad y un mal pronóstico.
Sin embargo, la mayoría de los casos de LMA presentan un riesgo intermedio cuando se clasifica utilizando criterios cito genéticos tradicionales. Estos casos tienen un cariotipo normal o anomalías cromosómicas que no están claramente asociadas con un pronóstico favorable o desfavorable. A pesar de ello, dentro de este grupo intermedio, existen variantes genéticas recurrentes que tienen relevancia pronóstica. Por ejemplo, la duplicación interna en tándem del gen FLT3 (FLT3-ITD), que se observa en aproximadamente el 30% de los casos de LMA, se asocia condicionalmente con un pronóstico pobre cuando está presente en el contexto de una mutación normal del gen NPM1 (nucleofosmina 1). Además, otras variantes patogénicas que también se asocian con un pronóstico desfavorable incluyen mutaciones en los genes RUNX1, ASXL1 y TP53, que alteran la función de genes cruciales para el control del ciclo celular y la reparación del ADN.
Por otro lado, existe un subgrupo relativamente favorable de pacientes que no presenta la mutación FLT3-ITD. Estos pacientes suelen portar variantes en el gen NPM1 o mutaciones bialélicas en el gen CEBPA (CCAAT/enhancer-binding protein alpha). Estas alteraciones están asociadas con una mejor respuesta al tratamiento y un pronóstico más favorable en comparación con otros subtipos de LMA.
Leucemia promielocítica aguda
La leucemia promielocítica aguda (LPA) es un subtipo específico de leucemia mieloide aguda que se discute por separado debido a sus características biológicas únicas y su respuesta excepcional a tratamientos que no son quimioterápicos, lo que la distingue de otros tipos de leucemia mieloide aguda. La LPA se caracteriza por una translocación cromosómica t(15;17), que da lugar a la formación de un gen de fusión denominado PML-RAR-alfa. Esta translocación implica una reordenación de los cromosomas 15 y 17, lo que provoca la fusión de dos genes clave: el gen PML (promyelocytic leukemia) y el gen RAR-alfa (receptor de ácido retinoico alfa). Esta alteración genética tiene un impacto profundo en la diferenciación de las células mieloides, causando un bloqueo en su maduración normal y dando lugar a la acumulación de promielocitos inmaduros en la médula ósea.
Una de las características más destacadas de la LPA es que, a pesar de ser una forma de leucemia aguda, tiene un pronóstico altamente favorable en comparación con otros subtipos de leucemia mieloide aguda. El tratamiento de la LPA ha experimentado una evolución significativa gracias a la introducción de terapias dirigidas que aprovechan la biología específica de la enfermedad. En particular, la integración de ácido todo-trans retinoico (ATRA) y trióxido de arsénico (ATO) en los regímenes de inducción, consolidación y mantenimiento ha transformado la LPA en una de las formas más curables de leucemia. Estos tratamientos no son quimioterápicos en el sentido tradicional, ya que actúan sobre las células leucémicas al restaurar la diferenciación celular y superar el bloqueo de maduración causado por la fusión del gen PML-RAR-alfa.
El ácido todo-trans retinoico (ATRA) es una forma activa de vitamina A que actúa como un agonista del receptor de ácido retinoico, el cual está afectado por la translocación cromosómica en la LPA. El ATRA se une al receptor RAR-alfa del gen de fusión PML-RAR-alfa y, al hacerlo, promueve la diferenciación de los promielocitos inmaduros en células mieloides maduras. Esto permite la normalización del proceso de maduración de las células sanguíneas, ayudando a la resolución de la leucemia. Por otro lado, el trióxido de arsénico (ATO) también tiene un efecto terapéutico sobre las células leucémicas de LPA al inducir la degradación del gen PML-RAR-alfa fusionado, restaurando así la función normal de las vías de diferenciación celular y contribuyendo a la eliminación de las células leucémicas.
La combinación de ATRA y ATO en el tratamiento de la LPA ha demostrado ser extraordinariamente efectiva, logrando tasas de curación superiores al 90% en muchos casos. Este éxito se debe a que estos tratamientos específicamente abordan la causa molecular de la enfermedad, superando la disfunción del proceso de diferenciación que caracteriza a la LPA. A diferencia de otras formas de leucemia mieloide aguda, donde la quimioterapia convencional es la piedra angular del tratamiento, la LPA puede ser tratada de manera muy eficaz con terapias dirigidas que no tienen los efectos secundarios tan devastadores de la quimioterapia tradicional.
El tratamiento de la LPA se realiza en fases de inducción, consolidación y mantenimiento. Durante la fase de inducción, se utiliza principalmente ATRA junto con ATO para inducir la diferenciación celular y reducir la carga leucémica. La fase de consolidación y mantenimiento incluye un tratamiento prolongado para asegurar la remisión completa de la enfermedad y prevenir la recurrencia, combinando ATRA y ATO, y en algunos casos, quimioterapia en dosis bajas. Esta estrategia terapéutica ha llevado a una mejora notable en la supervivencia a largo plazo de los pacientes con LPA.
Leucemia linfoblástica aguda
La leucemia linfoblástica aguda (LLA) es un tipo de leucemia caracterizada por la proliferación descontrolada de linfoblastos, que son precursores inmaduros de los linfocitos, las células del sistema inmunológico. La clasificación de la LLA se realiza en función del linaje celular afectado, siendo los principales subtipos los linfocitos B y T, que son los dos tipos principales de linfocitos en el cuerpo. Dentro de la categoría de LLA de células B, se distingue entre dos grupos con características genéticas diferentes: aquellos con cromosoma Filadelfia positivo y aquellos con cromosoma Filadelfia negativo.
El cromosoma Filadelfia es una translocación cromosómica específica, t(9;22), que da lugar a un gen de fusión BCR-ABL, asociado con una forma más agresiva de leucemia y un pronóstico generalmente peor. En cambio, la LLA de células B sin esta translocación (es decir, cromosoma Filadelfia negativo) tiene un pronóstico más favorable. En la LLA de células T, que también afecta principalmente a niños y adolescentes, las características genéticas específicas son diferentes y, aunque el pronóstico puede ser más variable, se sigue una clasificación similar.
La citogenética de la LLA es fundamental para determinar el pronóstico de la enfermedad, ya que las alteraciones cromosómicas tienen una relación directa con el comportamiento clínico y la respuesta al tratamiento. Se han identificado varias anomalías cromosómicas que se asocian con un mejor pronóstico, entre ellas la hiperdiploidía, que se refiere a la presencia de más de 50 cromosomas en las células leucémicas. Esta condición se observa en un porcentaje significativo de casos de LLA y se asocia especialmente con la ganancia de los cromosomas 4, 10 y 17. Los pacientes con hiperdiploidía suelen tener una mayor probabilidad de lograr una remisión completa y una supervivencia a largo plazo, lo que los coloca en un grupo de pronóstico favorable.
Otra anomalía cromosómica favorable es la translocación t(12;21), que produce el gen de fusión TEL-AML1. Esta alteración es común en la LLA pediátrica y se asocia con un curso clínico más favorable, con tasas altas de curación en los niños afectados. La presencia de esta translocación implica una alteración genética que, aunque transformadora, no confiere la agresividad de otras alteraciones más desfavorables. Por lo tanto, la LLA con esta translocación tiene una probabilidad significativamente mayor de ser tratada con éxito.
Por otro lado, existen varias alteraciones cromosómicas desfavorables que están asociadas con un pronóstico pobre. La hipodiploidía, que implica la presencia de menos de 44 cromosomas en las células leucémicas, está asociada con una menor tasa de respuesta al tratamiento y un riesgo más alto de recaída. Esta anomalía es un marcador de enfermedad más agresiva y es un factor predictivo de una mala evolución clínica. La translocación t(9;22), que produce el cromosoma Filadelfia, es otro marcador genético desfavorable en la LLA. Esta translocación genera el gen de fusión BCR-ABL, que resulta en una proteína quinasa de actividad anormalmente alta, promoviendo la proliferación de las células leucémicas y haciéndolas resistentes a ciertos tratamientos.
Otra alteración desfavorable en la LLA es la translocación t(4;11), que involucra el gen MLL (Mixed Lineage Leukemia) en el cromosoma 11q23. Esta translocación genera una fusión génica que interfiere con la regulación de la diferenciación celular y está asociada con un pronóstico más reservado, caracterizado por una mayor resistencia a los tratamientos convencionales. Además, la presencia de un cariotipo complejo, que implica más de cinco alteraciones cromosómicas diferentes, también está vinculada a una enfermedad más difícil de tratar y a un pronóstico muy desfavorable.
Leucemia aguda de linaje ambiguo
La leucemia aguda de linaje ambiguo (ALAL) es un subtipo raro y complejo de leucemia caracterizado por la presencia de células inmaduras, conocidas como blastos, que no muestran una diferenciación clara hacia los linajes mieloide o linfoide. En este tipo de leucemia, las células leucémicas pueden expresar antígenos característicos de ambos linajes, es decir, tanto mieloides como linfoides, lo que dificulta su clasificación en los subtipos tradicionales de leucemia mieloide aguda o leucemia linfoblástica aguda. Esta peculiaridad biológica da lugar a una clasificación que se considera un «linaje ambiguo», ya que las células tumorales no se ajustan claramente a los criterios de los linajes convencionales de glóbulos blancos.
La ALAL es un subtipo de leucemia extremadamente agresivo y, debido a la heterogeneidad de sus características biológicas y su resistencia al tratamiento estándar, se clasifica como un grupo de alto riesgo. El hecho de que las células leucémicas expresen características tanto de células mieloides como de células linfoides contribuye a la dificultad para tratar esta enfermedad, ya que las terapias estándar que se utilizan para los tipos de leucemia de linaje definido (como la leucemia mieloide aguda o la leucemia linfoblástica aguda) no son siempre efectivas en este caso.
Desde el punto de vista molecular, la presencia de células que combinan antígenos de ambos linajes sugiere que la ALAL podría estar relacionada con alteraciones genéticas complejas que afectan la regulación de la diferenciación celular. Estas alteraciones pueden ser responsables de la capacidad de las células leucémicas para escapar del proceso normal de maduración, lo que favorece su proliferación descontrolada.
El pronóstico para los pacientes con ALAL es muy pobre, y el tratamiento es mucho más desafiante en comparación con otros tipos de leucemia. Dado que las características biológicas de la ALAL son tan atípicas, los enfoques terapéuticos convencionales suelen ser menos efectivos, lo que hace que el tratamiento de esta leucemia requiera un enfoque especializado. En función de la limitada información disponible sobre el tratamiento de la ALAL, los regímenes utilizados son similares a los de la leucemia linfoblástica aguda, dada la similitud de algunas características biológicas de los blastos. Así, se emplea un régimen de quimioterapia tipo leucemia linfoblástica aguda, que incluye medicamentos dirigidos a erradicar las células leucémicas.
En aquellos pacientes que presentan una translocación t(9;22), que da lugar al cromosoma Filadelfia, se recomienda añadir un inhibidor de tirosina quinasa. Esta translocación es una alteración genética comúnmente observada en varios tipos de leucemias, incluida la leucemia mieloide crónica y la leucemia linfoblástica aguda, y produce una proteína quimérica (BCR-ABL) que aumenta la proliferación de las células leucémicas. El tratamiento con inhibidores de tirosina quinasa como el imatinib puede bloquear esta proteína anómala y mejorar la respuesta terapéutica.
Sin embargo, dado el curso clínico agresivo y la alta tasa de recaída asociada con la ALAL, muchos expertos en el tratamiento de leucemias recomiendan el trasplante de células madre hematopoyéticas alogénicas (trasplante de células madre de un donante compatible) como una opción terapéutica a considerar, especialmente después de un tratamiento de inducción que intente lograr una remisión completa. El trasplante de células madre hematopoyéticas ofrece la posibilidad de restaurar la función hematopoyética normal en pacientes cuyas médulas óseas han sido invadidas por células leucémicas y que, además, tienen un alto riesgo de recaída debido a las características genéticas complejas de la enfermedad.
Manifestaciones clínicas
La leucemia aguda es una enfermedad hematológica que se caracteriza por la rápida aparición de síntomas, lo que provoca que la mayoría de los pacientes presenten un cuadro clínico en un periodo de días o semanas. Los síntomas más comunes de la leucemia aguda incluyen sangrados y infecciones, que son consecuencia de las alteraciones en la médula ósea y la disminución de las células sanguíneas normales, como las plaquetas y los neutrófilos.
Uno de los principales síntomas es el sangrado, que generalmente ocurre debido a la trombocitopenia, es decir, una disminución en el número de plaquetas en la sangre. Esto puede llevar a manifestaciones de sangrado en la piel y en las superficies mucosas, como gingivorragia (sangrado de las encías), epistaxis (sangrado nasal) y menorragia (sangrado menstrual excesivo). En algunos casos menos comunes, el sangrado puede volverse generalizado y grave, lo que se observa en los pacientes que desarrollan coagulación intravascular diseminada (CID). Esta complicación, que es más frecuente en la leucemia promielocítica aguda y en la leucemia monocítica, se caracteriza por la formación de pequeños coágulos en los vasos sanguíneos, lo que puede consumir las plaquetas y los factores de coagulación, resultando en hemorragias masivas.
La infección es otro hallazgo frecuente en los pacientes con leucemia aguda y ocurre debido a la neutropenia, es decir, la disminución de los neutrófilos, que son las células encargadas de defender al cuerpo contra infecciones bacterianas. A medida que el recuento de neutrófilos disminuye por debajo de 500 células por microlitro de sangre (0.5 × 10^9/L), el riesgo de infecciones aumenta significativamente. Las infecciones más comunes en estos pacientes incluyen celulitis, neumonía e infecciones perirectales, y si el tratamiento con antibióticos apropiados se retrasa, la muerte puede ocurrir en pocas horas debido a la rápida progresión de la infección. Además, las infecciones fúngicas también son prevalentes en estos pacientes debido a la alteración de la función inmune.
Otro síntoma importante en los pacientes con leucemia aguda es el dolor en las encías y la hipertrofia gingival, que puede hacer que los pacientes busquen atención médica. Además, el dolor en huesos y articulaciones también es común, ya que la proliferación de células leucémicas en la médula ósea puede generar dolor óseo generalizado, especialmente en áreas como el esternón, la tibia y el fémur.
Una de las presentaciones más dramáticas de la leucemia aguda es la hiperleucocitosis, en la cual el número de blastos circulantes (células inmaduras) se eleva de manera significativa, superando los 100,000 células por microlitro de sangre (100 × 10^9/L). Este aumento en el número de células inmaduras puede alterar la circulación sanguínea, lo que resulta en síntomas como dolor de cabeza, confusión y dificultad para respirar. La hiperleucocitosis es una urgencia médica y requiere tratamiento inmediato con quimioterapia, y en algunos casos, se realiza un procedimiento denominado leucaféresis para reducir la cantidad de leucocitos en sangre. Si no se trata rápidamente, la mortalidad en las primeras 48 horas puede ser superior al 40%.
Al examen físico, los pacientes con leucemia aguda suelen presentarse pálidos y con purpura (manchas moradas en la piel causadas por sangrados pequeños) y petequias (pequeñas manchas rojas o moradas causadas por la ruptura de pequeños vasos sanguíneos). Aunque los signos de infección no siempre son evidentes, algunos pacientes pueden presentar estomatitis (inflamación y úlceras en la mucosa bucal) o hipertrofia gingival. En los pacientes con leucemia monocítica, también pueden observarse fisuras rectales, que son úlceras o desgarros en la mucosa del recto debido a la invasión de células leucémicas en los tejidos. Además, puede haber un aumento variable del tamaño del hígado, el bazo y los ganglios linfáticos, lo que indica una expansión de la enfermedad hacia otros órganos.
Exámenes diagnósticos
La característica distintiva de la leucemia aguda es la combinación de pancitopenia, que se refiere a la disminución de todas las líneas celulares sanguíneas (glóbulos rojos, glóbulos blancos y plaquetas), junto con la presencia de blastos circulantes en la sangre periférica. Los blastos son células inmaduras que proliferan de manera descontrolada y que son la base del diagnóstico de leucemia. Sin embargo, en algunos casos, los blastos pueden no estar presentes en el frotis sanguíneo periférico, lo que ocurre en aproximadamente el 10% de los casos de leucemia aguda, lo que se denomina «leucemia aleucémica». En estos casos, la ausencia de blastos en la sangre periférica no impide el diagnóstico, ya que estos blastos pueden estar presentes principalmente en la médula ósea o en otros tejidos.
La médula ósea, en la mayoría de los pacientes con leucemia aguda, suele ser hipercelular, lo que significa que está ocupada en su mayoría por células inmaduras (blastos), superando el umbral del 20% de células blasticas en el total de las células medulares. Este aumento en la cantidad de células leucémicas impide la producción normal de células sanguíneas, lo que resulta en pancitopenia. Además, se puede observar hiperuricemia, que es un aumento de los niveles de ácido úrico en la sangre. Este fenómeno ocurre debido a la destrucción rápida de las células leucémicas y la liberación masiva de productos de desecho celular, como las purinas, que se convierten en ácido úrico.
En aquellos casos donde está presente la coagulación intravascular diseminada (CID), una complicación grave asociada con algunas formas de leucemia, como la leucemia promielocítica aguda, se observan alteraciones en los parámetros de coagulación. En la CID, los niveles de fibrinógeno se reducen, el tiempo de protrombina se prolonga y los productos de degradación de la fibrina, o los dímeros D de fibrina, pueden ser detectados en la sangre, lo que indica un proceso de coagulación diseminada y un riesgo significativo de sangrados graves.
En algunos pacientes con leucemia linfoblástica aguda, especialmente en aquellos con leucemia de células T, puede observarse una masa mediastínica visible en la radiografía de tórax (radiografía de tórax), lo que sugiere la presencia de células leucémicas en el mediastino, el área entre los pulmones. Este hallazgo es más común en la leucemia de células T, que afecta principalmente a niños y adolescentes.
La leucemia meníngea es una complicación en la que las células leucémicas invaden el sistema nervioso central, y se puede detectar la presencia de blastos en el líquido cefalorraquídeo (LCR). Aunque esta complicación se observa en aproximadamente el 5% de los casos de leucemia aguda al momento del diagnóstico, es más frecuente en los subtipos monocíticos de leucemia mieloide aguda y también se puede observar en la leucemia linfoblástica aguda, especialmente en aquellos pacientes que no responden al tratamiento estándar.
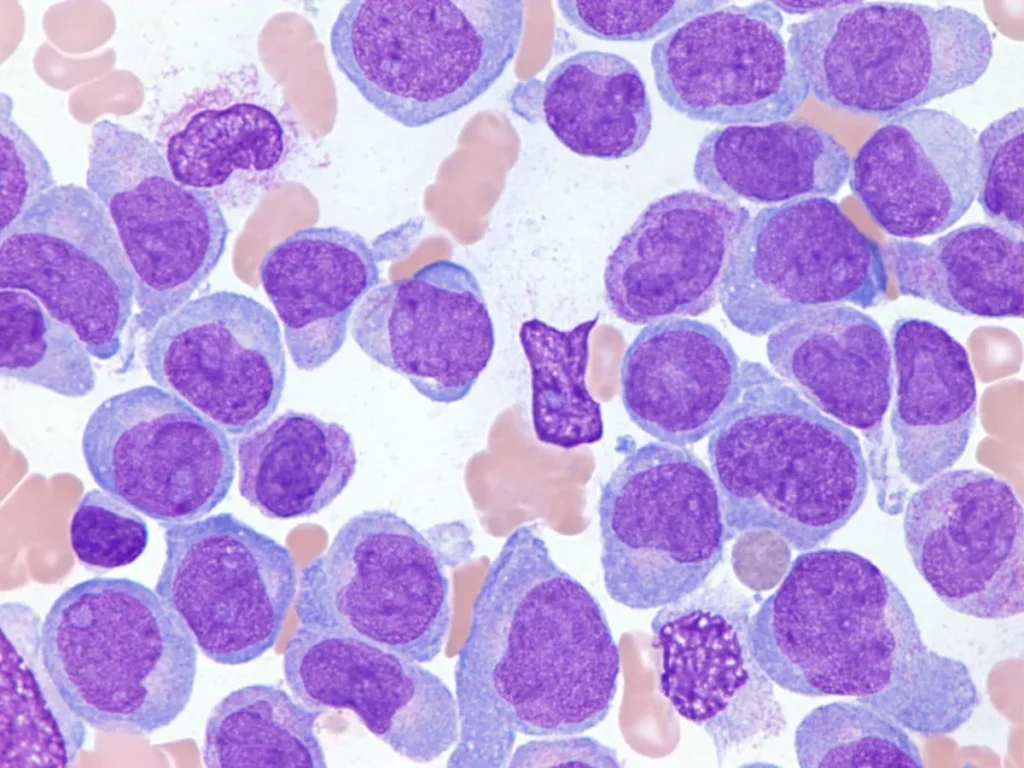
En la leucemia mieloide aguda, un hallazgo característico es la presencia de los «bastones de Auer», que son inclusiones citoplasmáticas eosinofílicas en forma de agujas. Estos bastones son una característica distintiva de la leucemia mieloide aguda, aunque también pueden encontrarse en otros trastornos hematológicos, como la leucemia promielocítica aguda, los síndromes mielodisplásicos de alto grado y los trastornos mieloproliferativos.
El fenotipo de las células leucémicas, que se refiere a sus características moleculares y antigénicas, generalmente se determina mediante técnicas de citometría de flujo o inmunohistoquímica. En la leucemia mieloide aguda, las células leucémicas suelen expresar antígenos mieloides, como CD13, CD33 y la mieloperoxidasa, una enzima típica de las células mieloides. En la leucemia linfoblástica aguda de linaje B, las células leucémicas suelen expresar el antígeno CD19, y la mayoría de los casos también expresan CD10, que se conoce como el «antígeno común de la leucemia linfoblástica aguda». En los casos de leucemia linfoblástica aguda de linaje T, las células leucémicas generalmente no expresan los marcadores T maduros, como CD3, CD4 o CD8, pero sí pueden expresar una combinación de marcadores como CD2, CD5 y CD7, y no expresan inmunoglobulina de superficie, lo que es característico de los linajes T inmaduros. Además, casi todas las células leucémicas, tanto de linaje mieloide como linfoide, expresan la terminal desoxirribonucleotidil transferasa (TdT), una enzima que es común en las células inmaduras.
Diagnóstico diferencial
La leucemia mieloide aguda (LMA) debe ser diferenciada de otros trastornos mieloproliferativos, como la leucemia mieloide crónica (LMC) y los síndromes mielodisplásicos (SMD), debido a las diferencias en sus características clínicas, citogenéticas y biológicas. Aunque todos estos trastornos afectan las células hematopoyéticas y pueden causar una proliferación anómala de las células sanguíneas, la LMA se caracteriza por la presencia de blastos inmaduros en la sangre periférica y en la médula ósea, lo que implica una interrupción significativa de la hematopoyesis normal. En cambio, en la LMC, la proliferación de células mieloides maduras y el hallazgo de la translocación del cromosoma Filadelfia (t(9;22)) son clave para el diagnóstico, mientras que en los síndromes mielodisplásicos existe una disfunción en la maduración celular que conduce a la producción de células hematopoyéticas defectuosas, pero no necesariamente a la proliferación de blastos.
La confusión entre la leucemia mieloide aguda y otros trastornos mieloproliferativos puede ser particularmente difícil cuando hay una proliferación de células inmaduras en la médula ósea, lo que puede hacer que la LMA se asemeje a una médula ósea en recuperación después de un daño tóxico previo. En estas situaciones, el diagnóstico puede ser incierto, y es necesario realizar un seguimiento estrecho con estudios adicionales. Si la leucemia mieloide aguda es sospechosa pero el diagnóstico no está claro, una estrategia útil consiste en repetir el estudio de la médula ósea después de unos días para observar si ha ocurrido una maduración de las células hematopoyéticas. En los casos de leucemia mieloide aguda, la proliferación de blastos sigue siendo predominante, sin que se observe una maduración significativa de las células, mientras que en la recuperación de la médula ósea tras un insulto tóxico, se esperaría ver un aumento progresivo de células maduras y la normalización de los parámetros hematológicos.
En cuanto a la leucemia linfoblástica aguda (LLA), su diagnóstico también puede ser desafiante debido a la necesidad de diferenciarla de otras enfermedades linfoides, como la leucemia linfocítica crónica (LLC), los linfomas y la leucemia de células peludas, entre otros trastornos. Estas enfermedades comparten ciertas características clínicas y hematológicas, como la presencia de linfocitos atípicos en la sangre periférica, pero se diferencian por su comportamiento biológico, las características de las células malignas y el curso de la enfermedad. En la leucemia linfoblástica aguda, los linfoblastos inmaduros invaden rápidamente la médula ósea, lo que interfiere con la producción de células sanguíneas normales, mientras que en la leucemia linfocítica crónica, los linfocitos anómalos suelen ser más maduros y el curso de la enfermedad es más indolente.
La leucemia linfoblástica aguda también puede confundirse con otras condiciones que presentan linfocitos atípicos en la sangre periférica, como la linfocitosis atípica en la mononucleosis infecciosa o la tos ferina. En la mononucleosis, causada por el virus de Epstein-Barr, se observa una proliferación de linfocitos atípicos, pero estos son generalmente reactivos y no malignos. La tos ferina, también conocida como pertussis, puede inducir una respuesta inflamatoria que provoca la aparición de linfocitos atípicos en la sangre. Aunque estas condiciones pueden compartir algunas características hematológicas con la leucemia linfoblástica aguda, la diferencia fundamental radica en la naturaleza reactiva de los linfocitos en estas infecciones, en contraste con los linfoblastos malignos característicos de la leucemia linfoblástica aguda.
Tratamiento
La leucemia aguda se considera una enfermedad curable, especialmente en pacientes más jóvenes que no tienen comorbilidades significativas. El primer paso en el tratamiento de la leucemia aguda es alcanzar una remisión completa. Esto se define como la normalización de la sangre periférica (es decir, que el recuento de las células sanguíneas vuelva a los niveles normales), la resolución de la citopenia (la disminución anormal de las células sanguíneas, como glóbulos rojos, glóbulos blancos y plaquetas), la normalización de la médula ósea (sin exceso de blastos, que son células inmaduras) y la restauración del estado clínico normal del paciente.
Una vez alcanzada la remisión completa, el siguiente paso es continuar con el tratamiento, que puede incluir consolidación y mantenimiento, dependiendo del tipo específico de leucemia. El tipo de quimioterapia inicial que se administra depende del subtipo de leucemia que se haya diagnosticado, ya que cada forma de leucemia tiene características biológicas y moleculares diferentes que requieren enfoques terapéuticos específicos.
La leucemia mieloide aguda (LMA) es una enfermedad hematológica agresiva que requiere un tratamiento intensivo, especialmente en pacientes más jóvenes sin comorbilidades significativas. El tratamiento de la LMA con intención curativa generalmente incluye una combinación de antraciclinas, como la daunorrubicina o la idarubicina, junto con citarabina, un agente quimioterapéutico que actúa sobre las células en fase de división rápida, que es característica de las células leucémicas. Esta combinación de fármacos ha demostrado ser eficaz, produciendo remisiones completas en el 80-90% de los pacientes menores de 60 años y en aproximadamente el 50-60% de los pacientes mayores. En algunos casos, la citarabina puede combinarse con otros agentes terapéuticos, como el gemtuzumab ozogamicina, un anticuerpo monoclonal conjugado con un agente quimioterapéutico, que mejora la respuesta en determinados subgrupos de pacientes.
Los pacientes con LMA secundaria, es decir, aquellos cuya enfermedad ha evolucionado a partir de trastornos mielodisplásicos o mieloproliferativos previos, o aquellos con LMA asociada al tratamiento de otras enfermedades, requieren un enfoque terapéutico diferente. Para estos pacientes, el tratamiento estándar incluye Vyxeos, una formulación liposomal que combina daunorrubicina y citarabina, diseñada para mejorar la entrega de los fármacos a las células leucémicas y reducir la toxicidad en los tejidos normales.
Un aspecto clave del tratamiento de la LMA es la identificación de variantes genéticas patogénicas que pueden influir en la respuesta a los tratamientos. Los pacientes con una mutación en el gen FLT3, que codifica una proteína tirosina quinasa involucrada en la proliferación celular, se benefician de la adición de midostaurina, un inhibidor de FLT3, a su régimen de tratamiento. Esta estrategia ha mostrado mejorar las tasas de remisión y las posibilidades de cura en estos pacientes.
El tratamiento post-remisión tiene como objetivo consolidar los beneficios obtenidos durante la fase inicial del tratamiento y evitar la recaída. Las opciones incluyen más quimioterapia o el trasplante de células madre hematopoyéticas alogénicas, es decir, trasplante de células madre de un donante compatible. Los pacientes con un perfil genético favorable pueden ser tratados con quimioterapia sola o con trasplante autólogo (trasplante con las propias células madre del paciente) y tienen una tasa de cura del 60-80%. Los pacientes con un perfil de riesgo intermedio tienen una tasa de cura del 35-40% con quimioterapia y un 40-60% con trasplante alogénico.
Por otro lado, los pacientes que no responden al tratamiento inicial o que tienen una genética de alto riesgo tienen tasas de cura inferiores al 10% con quimioterapia sola y son candidatos a trasplante de células madre alogénicas. Estos trasplantes de células madre alogénicas ofrecen una oportunidad de cura en aquellos pacientes que no alcanzan la remisión completa con la quimioterapia inicial.
En pacientes mayores de 75 años o con comorbilidades significativas, que no son candidatos a tratamiento con intención curativa, el tratamiento se enfoca en terapias dirigidas más específicas y menos intensivas. Estos agentes incluyen el inhibidor de BCL2 venetoclax, que se utiliza en combinación con un agente hipometilante o con citarabina a baja dosis, así como otros medicamentos dirigidos a mutaciones específicas, como enasidenib, que apunta a las mutaciones en el gen IDH2, e ivosidenib y olutasidenib, que se dirigen a las mutaciones en el gen IDH1. En algunos de estos pacientes, si logran un buen control de la enfermedad, el trasplante alogénico de intensidad reducida puede ofrecer beneficios adicionales.
Una vez que la leucemia ha recurrido después de la quimioterapia inicial, el pronóstico se vuelve más reservado. Para los pacientes que logran entrar en una segunda remisión, el trasplante alogénico de células madre sigue siendo la mejor opción, con una posibilidad de cura del 20-30%. Las terapias dirigidas mencionadas previamente pueden ser útiles en pacientes seleccionados y ofrecer un control de la enfermedad a largo plazo, aunque no garantizan la cura.
El tratamiento de la leucemia linfoblástica aguda en adultos se basa en la quimioterapia combinada, que incluye medicamentos como daunorrubicina, vincristina, prednisona y asparaginasa. Este enfoque terapéutico tiene como objetivo inducir una remisión completa en aproximadamente el 90% de los pacientes. La quimioterapia combinada actúa sobre las células leucémicas al interferir con su capacidad de dividirse y proliferar, lo que permite que el organismo recupere una hematopoyesis normal. Es importante destacar que el tratamiento para la leucemia linfoblástica aguda es generalmente menos mielosupresivo que el utilizado para la leucemia mieloide aguda, lo que significa que tiene un impacto menos negativo sobre la producción de células sanguíneas en la médula ósea, y no necesariamente provoca una aplasia medular prolongada.
Sin embargo, uno de los subtipos específicos de la leucemia linfoblástica aguda en adultos es la forma positiva para el cromosoma Filadelfia (o leucemia linfoblástica aguda positiva para bcr-abl). Este subtipo se caracteriza por una translocación cromosómica en la que los genes bcr y abl se fusionan, produciendo una proteína tirosina quinasa anómala que estimula la proliferación de células leucémicas. En estos pacientes, es fundamental agregar un inhibidor de la tirosina quinasa, como dasatinib o ponatinib, al tratamiento inicial, ya que estos medicamentos bloquean la actividad de la proteína bcr-abl y mejoran los resultados clínicos.
Un aspecto importante en el tratamiento de la leucemia linfoblástica aguda es la profilaxis del sistema nervioso central, ya que la leucemia puede infiltrarse en el cerebro y las meninges, lo que se conoce como secuestro meníngeo de células leucémicas. Para prevenir este fenómeno, se administran medicamentos intratecales (en el líquido cefalorraquídeo), como metotrexato o citarabina, durante el tratamiento.
Una vez que los pacientes alcanzan la remisión completa, se les puede administrar ciclos adicionales de quimioterapia para consolidar la remisión o, en algunos casos, quimioterapia a dosis altas seguida de un trasplante de células madre hematopoyéticas. La elección del tratamiento posterior depende de factores como la edad del paciente y el riesgo de la enfermedad. Los adultos más jóvenes, especialmente los menores de 39 años, suelen tener mejores resultados cuando se les trata con los protocolos diseñados para niños, que son más intensivos y específicos para este grupo de pacientes. Para los pacientes mayores, la prueba de enfermedad residual mínima (que detecta la presencia de células leucémicas que aún pueden estar presentes en la médula ósea después de la remisión) es crucial para identificar a aquellos con alto riesgo de recaída. En estos casos, el trasplante de células madre alogénicas (de un donante compatible) puede ser la opción de tratamiento más efectiva.
Para los pacientes cuya leucemia recurre, el tratamiento de segunda línea incluye terapias dirigidas más avanzadas. El anticuerpo biespecífico blinatumomab, que se dirige a las células leucémicas que expresan CD19, y el conjugado anticuerpo-fármaco inotuzumab ozogamicina, que se dirige a CD22, han demostrado una actividad notable y se consideran superiores a las opciones tradicionales de quimioterapia. De hecho, blinatumomab ha sido incorporado en el tratamiento de primera línea para los pacientes mayores y como enfoque de consolidación para todos los subgrupos de enfermedad. Esta terapia dirigida ha mostrado resultados prometedores, permitiendo un control más duradero de la enfermedad.
Además, la terapia con células T con receptores de antígenos quiméricos (CAR-T), que implica modificar las células T del paciente para que reconozcan y ataquen las células leucémicas que expresan CD19, ha sido aprobada para pacientes con leucemia linfoblástica aguda de tipo B recidivante o refractaria. Aunque actualmente se utiliza principalmente como puente para el trasplante de células madre alogénicas, la terapia con células CAR-T ha mostrado resultados impresionantes en términos de respuesta y curación en ciertos pacientes con leucemia linfoblástica aguda.
Pronóstico
La leucemia mieloide aguda en adultos tiene un pronóstico que varía significativamente según la edad del paciente y las características de la enfermedad. En los pacientes menores de 60 años, aproximadamente entre el 70% y el 80% logran una remisión completa con el tratamiento inicial, y de estos pacientes, alrededor del 50% pueden ser curados mediante terapias post-remisión adaptadas según el riesgo. El tratamiento post-remisión es crucial para consolidar la remisión alcanzada y prevenir la recaída, y puede incluir más ciclos de quimioterapia intensiva o el trasplante de células madre hematopoyéticas alogénicas, dependiendo del perfil genético de la leucemia y de la respuesta del paciente. Este enfoque dirigido ayuda a mejorar las tasas de cura, ya que se ajusta a las características específicas del paciente y de la enfermedad.
En contraste, los adultos mayores con leucemia mieloide aguda, especialmente aquellos mayores de 60 años, tienen un pronóstico menos favorable. Aunque alrededor del 50% de estos pacientes logran una remisión completa, las tasas de cura son significativamente más bajas, con apenas un 10% a un 20% de posibilidades de cura incluso si consiguen la remisión y reciben tratamiento post-remisión. Las razones de este menor índice de curación en pacientes mayores incluyen la menor capacidad de tolerar quimioterapia intensiva, la presencia de comorbilidades asociadas y una biología de la leucemia que puede ser más agresiva en este grupo etario.
Por otro lado, en los pacientes con leucemia linfoblástica aguda, aquellos menores de 39 años tienen un pronóstico mucho más favorable. Estos pacientes logran excelentes resultados después de recibir quimioterapia intensiva seguida de un tratamiento de intensificación adaptado al riesgo, que puede incluir trasplante de células madre hematopoyéticas. Las tasas de cura en este grupo de pacientes son del 60% al 80%, lo que refleja la mayor eficacia de los tratamientos agresivos en jóvenes y la menor incidencia de comorbilidades que podrían complicar el tratamiento.
Sin embargo, los pacientes con leucemia linfoblástica aguda que presentan citogenética adversa, una respuesta insuficiente a la quimioterapia o que son de edad avanzada tienen mucho menos chance de alcanzar una cura. Las tasas de cura en este grupo son considerablemente más bajas, entre el 20% y el 40%. La presencia de anomalías genéticas desfavorables, como las translocaciones cromosómicas específicas o las mutaciones en ciertos genes, puede hacer que la leucemia sea más resistente a los tratamientos estándar. En estos casos, se requieren enfoques terapéuticos más agresivos, que incluyen terapias dirigidas, trasplantes de células madre alogénicas o terapias experimentales, con el objetivo de mejorar las posibilidades de control a largo plazo.

Fuente y lecturas recomendadas:
- Advani AS et al. SWOG 1318: a phase II trial of blinatumomab followed by POMP maintenance in older patients with newly diagnosed Philadelphia chromosome-negative B-cell acute lymphoblastic leukemia. J Clin Oncol. 2022;40:1574. [PMID: 35157496]
- Curran E et al. Innovative approaches to the management of acute lymphoblastic leukemia across the age spectrum. Am Soc Clin Oncol Educ Book. 2022;42:1. [PMID: 35503981]
- Dholaria B et al. The evolving role of allogeneic haematopoietic cell transplantation in the era of chimaeric antigen receptor T-cell therapy. Br J Haematol. 2021;193:1060. [PMID: 33928630]
- El Chaer F et al. How I treat AML incorporating the updated classifications and guidelines. Blood. 2023;141:2813. [PMID: 36758209]
- Gupta S et al. Determinants of outcomes and advances in CD19-directed chimeric antigen receptor therapy for B-cell acute lymphoblastic leukemia. Eur J Haematol. 2024;112:51. [PMID: 38105391]
- McMurry H et al. IDH inhibitors in AML—promise and pitfalls. Curr Hematol Malig Rep. 2021;16:207. [PMID: 33939107]
- Sekeres MA et al. American Society of Hematology 2020 guidelines for treating newly diagnosed acute myeloid leukemia in older adults. Blood Adv. 2020;4:3528. [PMID: 32761235]
- Swaminathan M et al. Novel therapies for AML: a round-up for clinicians. Expert Rev Clin Pharmacol. 2020;13:1389. [PMID: 33412978]
Aprende administración paso a paso

ADMINISTRACION DESDE CERO
Originally posted on 19 de noviembre de 2024 @ 11:39 AM

