Prueba tu suerte 🍀 ¡COMPRA UN BOLETO!

La anemia de células falciformes es un trastorno autosómico recesivo que se caracteriza por la presencia de hemoglobina anormal en los glóbulos rojos (RBC, por sus siglas en inglés), lo que conduce a una hemólisis crónica y a una serie de consecuencias clínicas graves. Esta patología se origina a partir de una mutación puntual en el ADN, en la que un solo cambio en una base nitrogenada sustituye el ácido glutámico por la valina en la sexta posición de la cadena beta de la hemoglobina. Este cambio específico, conocido como la mutación de la hemoglobina S (HbS), da lugar a una forma anormal de la hemoglobina que es incapaz de llevar a cabo su función de transporte de oxígeno de manera eficiente, y tiene una tendencia a polymerizarse bajo condiciones fisiológicas que favorecen un ambiente de bajo oxígeno (hipoxemia) o acidosis.
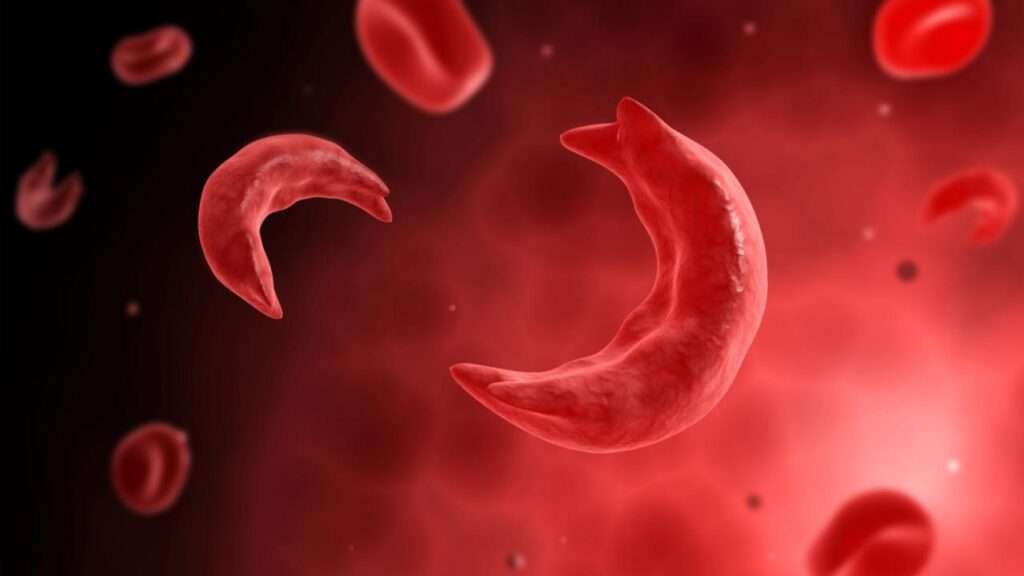
En condiciones normales, la hemoglobina está compuesta por dos cadenas alfa y dos cadenas beta, formando un tetrámero denominado hemoglobina normal (HbA), cuya principal función es transportar oxígeno desde los pulmones a los tejidos. Sin embargo, en la anemia de células falciformes, la presencia de hemoglobina S sustituye la hemoglobina normal, formando un tetrámero de dos cadenas alfa y dos cadenas beta S, conocido como hemoglobina SS. Esta hemoglobina S es inestable y, cuando se encuentra en un entorno con bajas concentraciones de oxígeno o con un pH ácido, tiende a polimerizarse, lo que provoca la deformación de los glóbulos rojos en una forma de media luna o falce. Este fenómeno es lo que da nombre a la enfermedad.
Las células falciformes tienen una vida útil significativamente más corta que los glóbulos rojos normales, ya que se destruyen más rápidamente en el proceso conocido como hemólisis. Esta destrucción de los glóbulos rojos libera hemoglobina libre en la circulación, lo que induce la liberación de ATP. El ATP liberado se convierte en adenosina, que se une a sus receptores específicos en las células del cuerpo. La adenosina, al unirse al receptor A2B, induce la producción de 2,3-bisfosfoglicerato (2,3-BPG), una molécula que favorece la formación de más polímeros de hemoglobina S y, por lo tanto, exacerba el proceso de falciformación. Además, la adenosina se une al receptor A2A en las células NK (células asesinas naturales), lo que conduce a la activación de una respuesta inflamatoria pulmonar.
Otro mecanismo patogénico importante es la captación de oxígeno por la hemoglobina falciforme, que no solo disminuye la capacidad de transporte de oxígeno en los tejidos, sino que también produce un fenómeno conocido como «escavenging» de óxido nítrico (NO). El óxido nítrico es una molécula importante en la regulación de la vasodilatación, pero cuando la hemoglobina S lo secuestra, se reduce su disponibilidad, lo que favorece la disfunción endotelial, el daño vascular y, en última instancia, la hipertensión pulmonar. Este proceso está relacionado con las complicaciones cardiovasculares y respiratorias que afectan a los pacientes con anemia de células falciformes.
La tasa de falciformación de los glóbulos rojos está influenciada por diversos factores, entre ellos la concentración intracelular de hemoglobina S y la presencia de otras variantes de hemoglobina en el interior de los glóbulos rojos. Una de las formas de hemoglobina que puede reducir significativamente la tasa de sickling es la hemoglobina fetal (HbF). Esta forma de hemoglobina tiene una afinidad más alta por el oxígeno y, lo más importante, no participa en la formación de polímeros de hemoglobina S. Por lo tanto, los pacientes con una mayor cantidad de hemoglobina fetal en la sangre experimentan una menor frecuencia de crisis de falcificación y una menor gravedad de la enfermedad.
Además, existen varios factores fisiológicos y patológicos que pueden aumentar la probabilidad de sickling en los glóbulos rojos. La deshidratación de los glóbulos rojos es un factor importante, ya que la pérdida de agua intracelular aumenta la concentración de hemoglobina S, favoreciendo la polimerización. Otros factores que incrementan el riesgo de sickling incluyen la acidosis, que reduce la afinidad de la hemoglobina S por el oxígeno, y la hipoxemia, una condición en la que los niveles de oxígeno en la sangre son anormalmente bajos. La hipoxia puede ser sistémica o localizada en los tejidos, lo que lleva a un aumento de la polimerización de la hemoglobina S y a la formación de células falciformes.
Las crisis hemolíticas en la anemia de células falciformes pueden estar relacionadas con varios factores. En los niños pequeños, antes de que el bazo sufra una infarto debido a la repetida falcificación de las células, se puede producir una seclusión esplénica, en la que los glóbulos rojos falciformes quedan atrapados en los vasos sanguíneos del bazo, lo que puede resultar en una crisis hemolítica aguda. Además, en pacientes con anemia de células falciformes, la presencia de trastornos concurrentes, como la deficiencia de glucosa-6-fosfato deshidrogenasa (G6PD), puede agravar las crisis hemolíticas y empeorar el pronóstico de la enfermedad.
Manifestaciones clínicas
La anemia de células falciformes tiene su inicio durante el primer año de vida, un momento crítico en el desarrollo hematológico del niño, que coincide con la caída de los niveles de hemoglobina fetal (hemoglobina F). La hemoglobina fetal es la forma predominante de hemoglobina en los recién nacidos y está compuesta por dos cadenas alfa y dos cadenas gamma, las cuales tienen una mayor afinidad por el oxígeno que las cadenas beta de la hemoglobina adulta. Esta hemoglobina se produce a través de un mecanismo genético que involucra la transcripción de la cadena gamma-globina. Sin embargo, hacia el final del primer año de vida, el organismo comienza a hacer una transición hacia la producción de hemoglobina adulta, en la cual las cadenas gamma son reemplazadas por las cadenas beta. Este cambio, denominado «switching» de globinas, es impulsado por señales genéticas que activan la transcripción de los genes que codifican la cadena beta-globina, específicamente la beta-globina que forma parte de la hemoglobina S en los individuos con anemia de células falciformes.
El descenso en los niveles de hemoglobina fetal en los primeros meses de vida y la progresiva sustitución por hemoglobina S es un evento crucial para la aparición de los síntomas clínicos característicos de la enfermedad. La hemoglobina S es inestable y tiene la tendencia a polimerizar bajo condiciones de hipoxia o acidosis, lo que da lugar a la deformación de los glóbulos rojos en forma de media luna o «falciforme». Esta deformación altera la estructura y la función de los glóbulos rojos, que se vuelven más frágiles y propensos a la hemólisis, lo que conduce a una anemia crónica hemolítica. La destrucción acelerada de los glóbulos rojos liberados de hemoglobina S causa un exceso de bilirrubina en la sangre, lo que se traduce en ictericia (color amarillo de la piel y las mucosas), que es un signo común de la enfermedad. Además, la bilirrubina no conjugada en la sangre se puede precipitar como cristales de calcio bilirrubinate, formando cálculos biliares pigmentados, un fenómeno frecuente en los pacientes con anemia de células falciformes.
La hemólisis crónica también puede provocar esplenomegalia, que es un agrandamiento del bazo debido a la acumulación de glóbulos rojos falciformes que son destruidos en ese órgano. Este fenómeno es especialmente evidente en los primeros años de vida, antes de que el bazo se infarte por la repetida obstrucción de sus vasos sanguíneos, una complicación que puede desarrollarse con el tiempo debido a la acumulación crónica de células falciformes. Además, los pacientes pueden presentar úlceras cutáneas, especialmente en la parte inferior de las piernas (tibia), que son difíciles de sanar. Estas úlceras son causadas por la microcirculación comprometida debido a la obstrucción de los pequeños vasos sanguíneos por los glóbulos rojos falciformes.
Las crisis hemolíticas agudas pueden poner en peligro la vida del paciente, especialmente durante los episodios de hemólisis o de crisis aplásicas. Las crisis hemolíticas agudas se desencadenan por diversos factores que aumentan la destrucción de los glóbulos rojos, como infecciones, deshidratación o hipoxia. La crisis aplásica, por otro lado, se asocia con una disminución temporal de la producción de glóbulos rojos en la médula ósea y está frecuentemente relacionada con infecciones virales (por ejemplo, el virus de parvovirus B19), que afectan la capacidad de la médula ósea para producir nuevas células sanguíneas. Además, los pacientes con anemia de células falciformes son particularmente vulnerables a la deficiencia de ácido fólico, un nutriente esencial para la producción de glóbulos rojos, lo que puede agravar la falta de producción de glóbulos rojos y contribuir al desarrollo de crisis aplásicas.
Los episodios dolorosos agudos, conocidos como «crisis vaso-oclusivas», son una manifestación clínica característica de la enfermedad y ocurren cuando los glóbulos rojos falciformes se agrupan y bloquean el flujo sanguíneo en los vasos pequeños. Estos episodios pueden ocurrir espontáneamente o ser provocados por factores como infecciones, deshidratación o hipoxia. Las áreas más comúnmente afectadas por las crisis vaso-oclusivas son la columna vertebral y los huesos largos de las extremidades, ya que son regiones del cuerpo con una microcirculación más densa y donde los glóbulos rojos falciformes tienen más probabilidades de obstruir los vasos sanguíneos. Durante estos episodios, los pacientes experimentan un dolor intenso que puede durar desde varias horas hasta varios días y, en muchos casos, puede ir acompañado de fiebre de bajo grado. Es importante señalar que los episodios vaso-oclusivos no están asociados con un aumento de la hemólisis, ya que el dolor proviene de la obstrucción de los vasos sanguíneos y la falta de oxígeno en los tejidos afectados, más que de la destrucción de los glóbulos rojos.
Uno de los eventos más graves que puede ocurrir durante una crisis vaso-oclusiva es un accidente cerebrovascular. Los episodios de vaso-oclusión pueden desencadenar trombosis en los senos venosos del cerebro (como la trombosis del seno sagital) o en las arterias cerebrales, lo que puede resultar en isquemia cerebral. Estos eventos pueden ser de naturaleza isquémica (bloqueo del flujo sanguíneo en los vasos) o hemorrágica (hemorragia secundaria a la ruptura de un vaso sanguíneo), ambos con consecuencias neurológicas potencialmente devastadoras. Además, las crisis vaso-oclusivas también pueden llevar a la aparición de priapismo, que es una erección dolorosa y persistente del pene, causada por la obstrucción de los vasos sanguíneos en la región pélvica.
La anemia de células falciformes es un trastorno hematológico caracterizado por la formación de glóbulos rojos falciformes, que tienen la capacidad de obstruir los pequeños vasos sanguíneos, lo que resulta en una serie de complicaciones clínicas graves. Los episodios repetidos de oclusión vascular, provocados por la polimerización de la hemoglobina S bajo condiciones de hipoxia o acidosis, afectan especialmente a órganos vitales como el corazón, los pulmones y el hígado, causando daño orgánico crónico y complicaciones que, en muchos casos, ponen en riesgo la vida del paciente.
Uno de los problemas más comunes y peligrosos en los pacientes con anemia de células falciformes es el síndrome torácico agudo. Este síndrome se caracteriza por la aparición repentina de dolor en el pecho, hipoxemia (niveles bajos de oxígeno en la sangre) y la presencia de infiltrados pulmonares visibles en una radiografía de tórax, lo que puede sugerir la presencia de una neumonía infecciosa. Sin embargo, es esencial distinguir entre el síndrome torácico agudo y una neumonía infecciosa, ya que ambos pueden presentar síntomas similares pero requieren tratamientos diferentes. El síndrome torácico agudo se asocia con la oclusión de los vasos pulmonares por los glóbulos rojos falciformes, lo que provoca una respuesta inflamatoria e insuficiencia respiratoria, y es una de las complicaciones respiratorias más graves de la enfermedad.
El daño isquémico a otros órganos, como los huesos, también es una característica prominente de la anemia de células falciformes. La necrosis isquémica de los huesos, particularmente de los huesos largos y de la cabeza de los fémures, puede ocurrir como resultado de la obstrucción de los vasos sanguíneos que irrigan estos tejidos. Esta necrosis aumenta la susceptibilidad de los huesos a infecciones bacterianas, particularmente aquellas causadas por Salmonella (un patógeno comúnmente asociado con osteomielitis en estos pacientes) y, con menor frecuencia, por Staphylococcus.
En el sistema renal, la oclusión de los vasos sanguíneos en la médula renal puede llevar a un infarto de las papilas renales, lo que ocasiona defectos en la capacidad de concentración de la orina por parte de los túbulos renales. Esto se traduce en una mayor pérdida de agua a través de la orina, lo que puede resultar en deshidratación. Además, la hematuria macroscópica (presencia de sangre en la orina) puede ser un signo visible de este daño renal, una manifestación que es más frecuente en individuos con rasgo de células falciformes (hemoglobina AS) que en aquellos con anemia de células falciformes en su forma homozygótica (hemoglobina SS).
Otro órgano frecuentemente afectado es la retina. La retinopatía en la anemia de células falciformes es común y puede progresar a una pérdida significativa de la visión si no se detecta y se maneja a tiempo. Los cambios retinianos incluyen la formación de neovasos, hemorragias y áreas de isquemia retinal, lo que puede llevar a complicaciones graves como el desprendimiento de retina y la ceguera.
La hipertensión pulmonar es otra complicación grave que puede desarrollarse en los pacientes con anemia de células falciformes, y se asocia con un pronóstico pobre. Esta condición ocurre debido a la obstrucción crónica de los vasos pulmonares por los glóbulos rojos falciformes, lo que aumenta la presión en la circulación pulmonar y puede llevar a insuficiencia cardíaca derecha. La hipertensión pulmonar es particularmente peligrosa en estos pacientes porque, al ser una enfermedad progresiva, puede causar un deterioro generalizado en la función cardiovascular.
La pubertad retrasada es un hallazgo común en los pacientes con anemia de células falciformes, probablemente debido a los efectos generales de la enfermedad sobre el crecimiento y el desarrollo, así como al impacto de la hipoxia crónica y los episodios de crisis dolorosa en la función hormonal y endocrina. Los adolescentes con esta enfermedad pueden experimentar un retraso en el inicio de la pubertad, lo que puede influir en su crecimiento físico y en el desarrollo de las características sexuales secundarias.
La susceptibilidad a infecciones es otra característica importante de la anemia de células falciformes. Los pacientes con esta enfermedad tienen un riesgo elevado de infecciones debido a la función reducida del bazo (hipoesplenismo). El bazo es un órgano crucial para la eliminación de bacterias y la respuesta inmune, y su función se ve comprometida en estos pacientes debido a la repetida obstrucción de los vasos esplénicos por los glóbulos rojos falciformes, lo que lleva a su infarto y fibrosis. Además, los defectos en la vía alternativa del complemento (un componente del sistema inmune) también contribuyen a una mayor vulnerabilidad a las infecciones, particularmente aquellas causadas por bacterias encapsuladas como Streptococcus pneumoniae.
En el examen físico, los pacientes con anemia de células falciformes suelen presentar una apariencia crónicamente enferma y, a menudo, una coloración amarillenta de la piel y las mucosas debido a la ictericia. El hígado también puede estar agrandado (hepatomegalia), lo que indica que está siendo sobrecargado por el trabajo de procesar los productos de la hemólisis, aunque la esplenomegalia (agrandamiento del bazo) es común en las etapas iniciales de la enfermedad, en la vida adulta es menos frecuente debido al infarto esplénico crónico. El corazón de los pacientes con anemia de células falciformes puede aparecer agrandado, y en algunos casos, se observa un precordio hiperactivo (aumento de la actividad cardíaca visible en la parte anterior del pecho) y murmullos sistólicos. Además, se puede detectar un aumento pronunciado en el segundo tono del corazón (P2), que refleja la presión elevada en la circulación pulmonar asociada con la hipertensión pulmonar.
Las úlceras cutáneas no cicatrizantes en la parte inferior de las piernas son comunes en los pacientes con anemia de células falciformes y se deben a la obstrucción crónica de los vasos sanguíneos en la piel, que compromete el suministro adecuado de oxígeno y nutrientes. Estas úlceras son difíciles de tratar y pueden ser una fuente de dolor y discapacidad para los pacientes.
Exámenes complementarios
La anemia hemolítica crónica es una característica fundamental de la anemia de células falciformes. El hematocrito de los pacientes con anemia de células falciformes suele estar entre el 20% y el 30%, lo que indica un nivel bajo de glóbulos rojos funcionales en la sangre. Este valor refleja la capacidad disminuida de la sangre para transportar oxígeno debido a la hemólisis constante. En el frotis de sangre periférica, los hallazgos característicos incluyen la presencia de células falciformes, que son glóbulos rojos deformados por la polimerización de hemoglobina S. Estas células falciformes pueden representar entre el 5% y el 50% de los glóbulos rojos en circulación, dependiendo de la gravedad de la enfermedad y de los episodios de crisis.
Además de las células falciformes, el frotis sanguíneo puede mostrar otras alteraciones que reflejan la respuesta del organismo a la hemólisis crónica. Una de estas es la reticulocitosis, que es el aumento en la cantidad de reticulocitos (glóbulos rojos inmaduros) en la sangre. Los reticulocitos son una respuesta a la necesidad de producir nuevos glóbulos rojos para compensar la destrucción prematura de los glóbulos rojos maduros. En la anemia de células falciformes, la reticulocitosis suele ser significativa, representando entre el 10% y el 25% del total de los glóbulos rojos. Además, debido a la hiperesplenismo inicial y la disfunción esplénica crónica, se pueden encontrar glóbulos rojos nucleados en la sangre periférica, un signo de que la médula ósea está liberando células inmaduras para satisfacer la demanda de glóbulos rojos.
Otro hallazgo importante en el frotis sanguíneo de pacientes con anemia de células falciformes es la presencia de cuerpos de Howell-Jolly y células en diana, que son indicativos de hipoesplenismo (función reducida o pérdida de la función del bazo). Los cuerpos de Howell-Jolly son fragmentos nucleares que normalmente son eliminados por el bazo en la circulación, pero que se encuentran en los pacientes con disfunción esplénica. Las células en diana, que son glóbulos rojos con un área de hemoglobina central rodeada por un anillo de hemoglobina, también son típicas en este contexto, reflejando alteraciones en la membrana celular de los glóbulos rojos debido a la hemólisis crónica.
En cuanto a los leucocitos, la recuento de glóbulos blancos (leucocitos) en la sangre periférica suele estar elevado, con un rango de 12,000 a 15,000 células por microlitro de sangre (12–15 × 10^9/L). Esta leucocitosis es reactiva y refleja una inflamación crónica debido a la hemólisis continua y a los episodios vaso-oclusivos que pueden desencadenar una respuesta inmune. Además, es frecuente observar trombocitosis reactiva, que es un aumento en el número de plaquetas, como parte de la respuesta inflamatoria y la reparación de los vasos sanguíneos dañados.
Los niveles de bilirrubina indirecta (no conjugada) suelen estar elevados debido a la destrucción constante de glóbulos rojos. La bilirrubina es un producto del catabolismo de la hemoglobina liberada durante la hemólisis, y su acumulación puede dar lugar a ictericia, que es un signo clínico visible de la enfermedad.
El diagnóstico definitivo de la anemia de células falciformes se realiza mediante electroforesis de hemoglobina, una prueba que permite identificar las diferentes formas de hemoglobina presentes en la sangre. En los pacientes con anemia de células falciformes, hemoglobina S será la principal forma de hemoglobina, representando entre el 85% y el 98% del total de hemoglobina. En la forma homozygótica de la enfermedad (anemia de células falciformes clásica), no se detecta hemoglobina A (la hemoglobina normal), ya que la producción de hemoglobina S es dominante. En algunos casos, se puede observar un aumento en los niveles de hemoglobina fetal (hemoglobina F), que es la forma de hemoglobina presente durante la vida fetal y en los recién nacidos. La presencia de hemoglobina F puede ser más pronunciada en algunos pacientes y se asocia con una evolución clínica más benigna, ya que la hemoglobina F no participa en la polimerización de hemoglobina S y reduce la tendencia a la formación de células falciformes. Niveles de hemoglobina F entre el 15% y el 20% están relacionados con un curso clínico menos severo y una menor frecuencia de crisis dolorosas.
Los pacientes con anemia de células falciformes y beta-talassemia positiva (S-beta+ talasemia) o con anemia de células falciformes y alfa-talassemia (SS alfa-talassemia) también tienden a tener una forma más benigna de la enfermedad. La beta-talassemia y la alfa-talassemia son trastornos genéticos que afectan la producción de las cadenas de hemoglobina, y la coexistencia de estas variantes con hemoglobina S modula la gravedad de los síntomas. En estos casos, la presencia de otras formas de hemoglobina, como la hemoglobina A o la hemoglobina F, contribuye a una mayor producción de hemoglobina funcional, lo que disminuye la frecuencia y la gravedad de las crisis vaso-oclusivas y otras complicaciones asociadas con la anemia de células falciformes.
Tratamiento
El tratamiento de la anemia de células falciformes se basa principalmente en el cuidado de apoyo, que tiene como objetivo aliviar los síntomas, prevenir complicaciones y mejorar la calidad de vida del paciente. La gestión integral de la enfermedad abarca una serie de intervenciones que van desde la suplementación de nutrientes esenciales hasta el uso de transfusiones y terapias farmacológicas para controlar las crisis agudas y prevenir las complicaciones crónicas.
Una de las medidas básicas en el manejo de la anemia de células falciformes es la suplementación con ácido fólico, que se administra generalmente a una dosis de 1 miligramo por vía oral diaria. El ácido fólico es esencial para la producción de glóbulos rojos, ya que favorece la síntesis de ADN y la maduración celular, contribuyendo a la compensación de la hemólisis crónica que caracteriza a esta enfermedad.
Cuando los pacientes experimentan crisis hemolíticas o crisis aplásicas (que implican una disminución temporal en la producción de glóbulos rojos debido a diversos factores, como infecciones virales o deficiencias nutricionales), se recurre a las transfusiones sanguíneas como tratamiento de apoyo para restablecer la cantidad y funcionalidad de los glóbulos rojos. Las transfusiones también se emplean en casos de anemia grave que no responden a otras medidas y pueden ser necesarias para prevenir complicaciones graves.
En cuanto a los episodios dolorosos agudos (crisis vaso-oclusivas), que son eventos caracterizados por el bloqueo de los vasos sanguíneos debido a la acumulación de glóbulos rojos falciformes, el tratamiento debe incluir la identificación de los factores precipitantes, como infecciones, deshidratación, hipoxia o cambios en el pH sanguíneo. Además, es crucial mantener a los pacientes bien hidratados, administrar analgésicos generosos para controlar el dolor, y proporcionar oxígeno suplementario si el paciente presenta hipoxia (niveles bajos de oxígeno en la sangre).
La prevención de infecciones es otra prioridad en el manejo de la anemia de células falciformes, ya que los pacientes son más susceptibles a infecciones debido a la función reducida del bazo (hipoesplenismo). Una de las intervenciones clave es la vacunación contra el neumococo, que ayuda a reducir la incidencia de infecciones graves causadas por esta bacteria, un patógeno particularmente peligroso en personas con anemia de células falciformes.
Además, existen tratamientos farmacológicos que han demostrado ser efectivos en la reducción de hospitalizaciones y en la mejora de la calidad de vida de los pacientes. La hidroxiurea es un medicamento que se utiliza para disminuir la frecuencia de las crisis dolorosas agudas y las hospitalizaciones relacionadas, al aumentar la producción de hemoglobina fetal, que no participa en la polimerización de la hemoglobina S, lo que disminuye la formación de glóbulos rojos falciformes. De manera similar, el L-glutamina, un aminoácido que tiene propiedades antioxidantes y antiinflamatorias, también ha mostrado eficacia en la reducción de crisis dolorosas agudas y hospitalizaciones, mejorando la función redox de los glóbulos rojos y protegiéndolos del daño.
En pacientes con microalbuminuria (presencia de pequeñas cantidades de proteína en la orina, un signo temprano de daño renal), se recomienda el uso de inhibidores de la enzima convertidora de angiotensina (ACE). Estos medicamentos tienen un efecto protector sobre los riñones al reducir la presión arterial y la sobrecarga de trabajo del sistema cardiovascular, lo que ayuda a prevenir la progresión de la insuficiencia renal.
En casos graves de crisis vaso-oclusivas o síndrome torácico agudo, o en situaciones donde se presenta priapismo o accidente cerebrovascular, las transfusiones de intercambio son una intervención crítica. Estas transfusiones consisten en reemplazar una parte significativa de los glóbulos rojos del paciente con sangre normal, lo que mejora rápidamente la oxigenación de los tejidos y reduce la obstrucción vascular. Las transfusiones de intercambio son particularmente útiles en crisis que no responden a otros tratamientos, como las que involucran órganos vitales como el cerebro, los pulmones y los genitales.
El tratamiento con transfusiones crónicas ha demostrado ser eficaz para reducir el riesgo de accidente cerebrovascular recurrente en niños con anemia de células falciformes. Se recomienda que los niños con la enfermedad sean sometidos a ultrasonidos transcraneales anuales para monitorear la velocidad de la sangre en los vasos cerebrales. Si la velocidad Doppler en estos vasos es anormal (superior a 200 cm/s), el tratamiento con transfusiones debe considerarse de manera preventiva para evitar un accidente cerebrovascular.
Sin embargo, las transfusiones crónicas conllevan el riesgo de sobrecarga de hierro, por lo que los pacientes que reciben transfusiones regulares requieren tratamiento de quelación de hierro. Este tratamiento tiene como objetivo eliminar el exceso de hierro del organismo, que se acumula debido a las transfusiones repetidas, y prevenir daños a órganos como el corazón y el hígado.
En lo que respecta al tratamiento de la inmunización contra los glóbulos rojos, se recomienda el uso de glóbulos rojos transfundidos que estén fenotípicamente emparejados con los del paciente para reducir el riesgo de aloimmunización. La aloimmunización ocurre cuando el sistema inmunológico del paciente forma anticuerpos contra los glóbulos rojos transfundidos, lo que puede complicar futuras transfusiones.
El tratamiento farmacológico de la anemia de células falciformes ha avanzado significativamente en los últimos años, ofreciendo a los pacientes diversas opciones terapéuticas que modulan la gravedad de la enfermedad y mejoran la calidad de vida. Las estrategias actuales se centran en aumentar los niveles de hemoglobina fetal, reducir la polimerización de la hemoglobina S y disminuir las crisis vaso-oclusivas recurrentes, que son las principales causas de morbilidad y complicaciones graves.
Una de las opciones más ampliamente utilizadas es la hidroxiurea, un agente farmacológico que aumenta los niveles de hemoglobina fetal de manera epigenética. La hidroxiurea actúa modificando la expresión genética de las células madre hematopoyéticas, estimulando la producción de hemoglobina F, que no participa en la polimerización de la hemoglobina S, reduciendo así la formación de células falciformes. La administración oral de hidroxiurea, con una dosis habitual de entre 15 y 35 mg por kilogramo de peso corporal al día, ha demostrado ser eficaz para reducir la frecuencia de las crisis dolorosas (crisis vaso-oclusivas) en pacientes cuya calidad de vida se ve gravemente afectada por episodios frecuentes de dolor, definidos como tres o más episodios al año. El seguimiento a largo plazo de los pacientes tratados con hidroxiurea ha mostrado una mejora en la supervivencia general y la calidad de vida, sin una evidencia significativa de que este tratamiento aumente el riesgo de malignidades secundarias, lo que lo convierte en una opción segura y beneficiosa para el manejo de la enfermedad.
Otra intervención farmacológica que ha mostrado beneficios es la suplementación con ácidos grasos omega-3 (ácidos grasos n-3), los cuales tienen propiedades antiinflamatorias. Estos suplementos pueden reducir las crisis vaso-oclusivas y disminuir la necesidad de transfusiones en pacientes con anemia de células falciformes. Aunque la evidencia es aún limitada en comparación con otros tratamientos, los ácidos grasos omega-3 constituyen una opción adicional que podría complementar el tratamiento estándar.
El L-glutamina es otro tratamiento que ha demostrado ser efectivo para modular favorablemente las crisis dolorosasasociadas con la anemia de células falciformes, así como el síndrome torácico agudo. El L-glutamina actúa protegiendo los glóbulos rojos falciformes del daño oxidativo y mejora la funcionalidad de los glóbulos rojos en circulación. Esto resulta en una reducción tanto de las crisis dolorosas como de la severidad de otros episodios complicados, mejorando así la calidad de vida de los pacientes.
En cuanto a los tratamientos más innovadores, se encuentra el anticuerpo monoclonal crizanlizumab-tmca, que ha demostrado reducir en un 50% las crisis vaso-oclusivas. Este medicamento actúa bloqueando la P-selectina, una molécula que se encuentra en las células endoteliales activadas. Al inhibir la interacción entre plaquetas, glóbulos rojos y leucocitos con el endotelio vascular, crizanlizumab interrumpe el proceso de obstrucción microvascular que caracteriza a las crisis vaso-oclusivas, proporcionando una mejora significativa en los episodios dolorosos y otras complicaciones asociadas con la enfermedad.
Otra opción terapéutica emergente es Voxelotor, un medicamento que inhibe la polimerización de la hemoglobina S desoxigenada. Al evitar que los glóbulos rojos falciformes se deformen bajo condiciones de hipoxia, voxelotor mejora la viscosidad sanguínea y aumenta los niveles de hemoglobina total en pacientes con anemia de células falciformes de 12 años o más. Esta mejora en los niveles de hemoglobina puede reducir significativamente la necesidad de transfusiones sanguíneas, lo que es particularmente beneficioso para aquellos pacientes que requieren transfusiones frecuentes debido a la gravedad de la enfermedad.
Existen tratamientos avanzados basados en la manipulación genética de células madre hematopoyéticas. Dos productos aprobados en los Estados Unidos para pacientes con enfermedad grave son lovotibeglogene autotemcel y exagamglogene autotemcel, que utilizan tecnologías avanzadas para modificar genéticamente las células madre hematopoyéticas del paciente. Lovotibeglogene autotemcel introduce una hemoglobina anti-falciforme (hemoglobina AT87Q), mientras que exagamglogene autotemcel utiliza la tecnología de edición genética CRISPR para aumentar los niveles de hemoglobina fetal, lo que también reduce la polimerización de hemoglobina S. Estas terapias génicas ofrecen la posibilidad de un control a largo plazo de la enfermedad y podrían representar una opción curativa para algunos pacientes con formas graves de la anemia de células falciformes.
Pronóstico
La anemia de células falciformes es una enfermedad crónica que afecta múltiples sistemas del cuerpo, lo que lleva a un deterioro progresivo de la función de varios órganos y a una reducción en la esperanza de vida. Desde que se describió la enfermedad por primera vez, el tratamiento ha mejorado significativamente, lo que ha permitido un aumento en la esperanza de vida de los pacientes. Sin embargo, la enfermedad sigue siendo una de las principales causas de morbilidad y mortalidad, especialmente cuando las complicaciones no se controlan adecuadamente.
La anemia hemolítica crónica que caracteriza a la anemia de células falciformes es una de las causas fundamentales del daño progresivo a los órganos. La hemólisis constante de los glóbulos rojos falciformes provoca una sobrecarga de bilirrubina, que contribuye a la aparición de ictericia y a la formación de cálculos biliares, lo que puede afectar el funcionamiento del hígado. La constante destrucción de glóbulos rojos también pone en marcha mecanismos compensatorios que a menudo no logran mantener una producción adecuada de sangre, lo que favorece la aparición de anemia grave.
La crisis vaso-oclusiva —debido a la acumulación de glóbulos rojos falciformes en los vasos sanguíneos pequeños— es una de las complicaciones más dolorosas y debilitantes, y puede ocurrir de manera repetida a lo largo de la vida de un paciente. Estas crisis, al ser frecuentes y severas, contribuyen al daño crónico de los órganos afectados, incluidos el corazón, los pulmones, el riñón y el cerebro. El daño endotelial, la hipertensión pulmonar, la infarctación renal y la accidente cerebrovascular son solo algunas de las consecuencias a largo plazo de las obstrucciones vasculares recurrentes.
Los pacientes con anemia de células falciformes también son particularmente vulnerables a las infecciones debido a la pérdida de la función esplénica, un fenómeno conocido como hipoesplenismo. El bazo, que desempeña un papel fundamental en la filtración de bacterias y en la respuesta inmunitaria, sufre infartos recurrentes debido a la obstrucción de los vasos sanguíneos que lo irrigan. Como resultado, la incidencia de infecciones graves, como las causadas por el neumococo, es considerablemente mayor en estos pacientes, lo que puede contribuir a un deterioro generalizado de la salud.
La enfermedad también puede dar lugar a complicaciones como hipertensión pulmonar, daño ocular (retinopatía) que puede llevar a pérdida de visión, y a úlceras cutáneas que no cicatrizan adecuadamente, especialmente en la región inferior de las piernas. Las complicaciones cardíacas, como la insuficiencia cardíaca o el infarto de miocardio, también son más comunes en los pacientes con anemia de células falciformes, debido a los efectos adversos de la hipoxia crónicay la hiperviscosidad sanguínea sobre el sistema cardiovascular.
A pesar de estas complicaciones, los avances en cuidado de apoyo, como el uso de transfusiones sanguíneas, el tratamiento con hidroxiurea, la suplementación con ácido fólico, y las terapias para reducir el dolor y mejorar la oxigenación, han aumentado notablemente la supervivencia de los pacientes con anemia de células falciformes. Con un tratamiento adecuado, la expectativa de vida promedio para los pacientes con esta enfermedad ha aumentado, y actualmente se estima que es de 40 a 50 años. Esta mejora en la esperanza de vida refleja no solo el mejor manejo de las crisis agudas y las complicaciones, sino también el acceso más amplio a tratamientos preventivos, como la vacunación y el uso de medicamentos modificadores de la enfermedad.
Aunque la esperanza de vida ha mejorado, sigue existiendo una reducción en la calidad de vida para muchos pacientes debido a la cronicidad de la enfermedad y la prevalencia de complicaciones graves. La investigación continúa en la búsqueda de tratamientos curativos y opciones que puedan detener o revertir el daño orgánico a largo plazo. En particular, las terapias genéticas y las transfusiones de células madre presentan un gran potencial para modificar el curso de la enfermedad y ofrecer una posibilidad de cura para algunos pacientes. Sin embargo, a pesar de estos avances, los pacientes con anemia de células falciformes continúan enfrentando desafíos significativos en términos de calidad de vida y supervivencia a largo plazo.

Fuente y lecturas recomendadas:
- DeBaun MR, et al. American Society of Hematology 2020 guidelines for sickle cell disease: prevention, diagnosis, and treatment of cerebrovascular disease in children and adults. Blood Adv. 2020; 4:1554. [PMID: 32298430]
- Esrick EB, et al. Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease. N Engl J Med. 2021; 384:205. [PMID: 33283990]
- Howard J, et al. Voxelotor in adolescents and adults with sickle cell disease (HOPE): long-term follow-up results of an international, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Haematol. 2021; 8. [PMID: 33838113]
- Kanter J, et al. Biologic and clinical efficacy of LentiGlobin for sickle cell disease. N Engl J Med. 2022; 386:617. [PMID: 34898139]
- Pecker LH, et al. Sickle cell disease. Ann Intern Med. 2021; 174. [PMID: 33428443]
Aprende administración paso a paso

ADMINISTRACION DESDE CERO
Originally posted on 17 de noviembre de 2024 @ 10:11 AM