El síndrome de Turner constituye un grupo de trastornos genéticos que afectan al cromosoma X y que están asociados con una serie de manifestaciones clínicas y biológicas complejas. Se trata de una condición que incide exclusivamente en individuos de sexo femenino y que se caracteriza principalmente por la ausencia parcial o total de un segundo cromosoma sexual, lo cual conlleva una serie de alteraciones del desarrollo somático y gonadal. Este síndrome es paradigmático dentro de las anomalías cromosómicas humanas, ya que pone de manifiesto la importancia del complemento cromosómico completo para el desarrollo normal.
Desde el punto de vista citogenético, aproximadamente el 50% de las pacientes con síndrome de Turner presentan un cariotipo 45,XO, es decir, carecen completamente de uno de los dos cromosomas X habituales. Esta monosomía implica la pérdida de material genético esencial que participa en el desarrollo embrionario, el crecimiento y la función reproductiva. El déficit de genes que normalmente se expresan a partir del segundo cromosoma X no se compensa adecuadamente, lo que se traduce en una serie de alteraciones fenotípicas distintivas. Entre las manifestaciones más comunes se encuentran la baja estatura, el desarrollo incompleto de los caracteres sexuales secundarios (hipogonadismo primario), la infertilidad, y anomalías morfológicas como el cuello alado, el tórax en escudo, la linfedema congénito y alteraciones cardiovasculares, como la coartación de la aorta.
El impacto del síndrome de Turner sobre la viabilidad fetal es notablemente alto. Se estima que afecta entre el 1% y el 2% de todos los embarazos, pero la gran mayoría de los fetos afectados, alrededor del 97%, no sobrevive hasta el nacimiento, lo que lo convierte en una de las causas cromosómicas más frecuentes de aborto espontáneo. De hecho, se calcula que aproximadamente un 10% de todas las pérdidas gestacionales espontáneas están asociadas a esta anomalía cromosómica. Esta elevada tasa de letalidad intrauterina refleja la incompatibilidad con la vida que genera, en muchos casos, la ausencia del cromosoma X adicional o la presencia de formas aberrantes del mismo.
A pesar de esta elevada tasa de pérdida prenatal, el síndrome de Turner puede observarse en aproximadamente 1 de cada 2500 nacimientos vivos de niñas. Esta supervivencia puede atribuirse, en muchos casos, a la presencia de mosaicismo, es decir, a la coexistencia de dos o más líneas celulares con diferente constitución genética. Algunas pacientes presentan células con cariotipo normal (46,XX) junto a células monosómicas (45,XO), mientras que otras pueden tener deleciones parciales del cromosoma X, cromosomas X en anillo, o incluso secuencias del cromosoma Y en algunas células. De hecho, alrededor del 12% de las pacientes con este síndrome presentan mosaicismo que incluye material del cromosoma Y, lo cual conlleva un riesgo aumentado de desarrollo de gonadoblastoma, un tipo de tumor germinal, si no se realiza una gonadectomía profiláctica.
Manifestaciones clínicas
El síndrome de Turner clásico, caracterizado citogenéticamente por la monosomía del cromosoma X (cariotipo 45,XO), representa una forma de disgenesia gonadal en la cual existe un fallo primario del desarrollo de las gónadas. Este trastorno tiene un espectro clínico notablemente amplio, cuyas manifestaciones varían tanto en severidad como en presentación temporal, desde la vida intrauterina hasta la adultez.
En el periodo neonatal, algunas niñas afectadas pueden ser identificadas desde el nacimiento debido a la presencia de hallazgos físicos evidentes. Entre los signos más prominentes se encuentra el linfedema congénito de manos y pies, resultado de una alteración en el desarrollo del sistema linfático. Estas recién nacidas también tienden a tener una talla menor a la esperada para la edad gestacional, lo que puede generar sospechas clínicas tempranas. Sin embargo, en muchas ocasiones el diagnóstico se retrasa hasta la infancia, cuando la principal causa de consulta suele ser la talla baja persistente. Este signo es una característica cardinal del síndrome de Turner, reflejo de la deficiencia de la hormona de crecimiento y de la disfunción gonadal subyacente, que afecta los picos puberales de crecimiento.
En la adolescencia, el hipogonadismo primario se manifiesta típicamente como un retraso puberal. En aproximadamente el ochenta por ciento de los casos, las niñas presentan amenorrea primaria, es decir, la ausencia completa del inicio de la menstruación. En el veinte por ciento restante, puede observarse un inicio puberal parcial seguido por una falla ovárica precoz. Esta variabilidad fenotípica se explica, en gran parte, por la presencia de mosaicismo genético: algunas pacientes que muestran signos de desarrollo puberal poseen poblaciones celulares con cariotipo mixto, por ejemplo 45,XO/46,XX, lo cual les permite una función ovárica parcial. La karyotipificación en sangre periférica puede no detectar este mosaicismo si está confinado a tejidos distintos, como los ovarios, lo que subraya la complejidad diagnóstica en casos con fenotipos atenuados.
En la adultez, el síndrome de Turner se caracteriza por una serie de rasgos físicos distintivos. Además de la baja estatura y el hipogonadismo, son comunes el cuello alado (una forma de pterigium colli), el paladar ojival, los pezones ampliamente espaciados, y otras dismorfias menores. Estas características se presentan con mayor frecuencia en pacientes con el fenotipo clásico (45,XO) que en aquellas con formas mosaico, cuyas manifestaciones pueden ser más sutiles.
Las alteraciones cardiovasculares representan un componente clínico de gran relevancia. La coartación de la aorta y la válvula aórtica bicúspide son dos de las anomalías más frecuentes y clínicamente significativas, asociadas a un aumento del riesgo de hipertensión arterial, disección aórtica y muerte súbita. Estas malformaciones se observan con mayor frecuencia en pacientes con cuello alado, lo que sugiere una posible correlación entre ciertas características dismórficas externas y defectos del desarrollo mesodérmico interno.
El compromiso sistémico del síndrome de Turner se extiende también al sistema renal, con frecuencia se detectan malformaciones como riñón en herradura o duplicación pieloureteral, lo que contribuye a un mayor riesgo de infecciones urinarias y disfunción renal a largo plazo. Además, las mujeres con este síndrome tienen una mayor susceptibilidad a enfermedades autoinmunes, siendo las más comunes la tiroiditis autoinmune (particularmente la enfermedad de Hashimoto), la enfermedad celíaca y, en menor medida, las enfermedades inflamatorias intestinales. Esta predisposición sugiere un papel regulador del cromosoma X en la tolerancia inmunológica.
En el ámbito neuroconductual, se ha documentado una mayor prevalencia de trastornos emocionales y dificultades en el procesamiento visoespacial, aunque la inteligencia general suele estar dentro de los parámetros normales. La vulnerabilidad psicológica se acentúa en la adolescencia, especialmente cuando las diferencias físicas y los problemas de fertilidad comienzan a tener implicaciones psicosociales.

Características de la cabeza y cuello
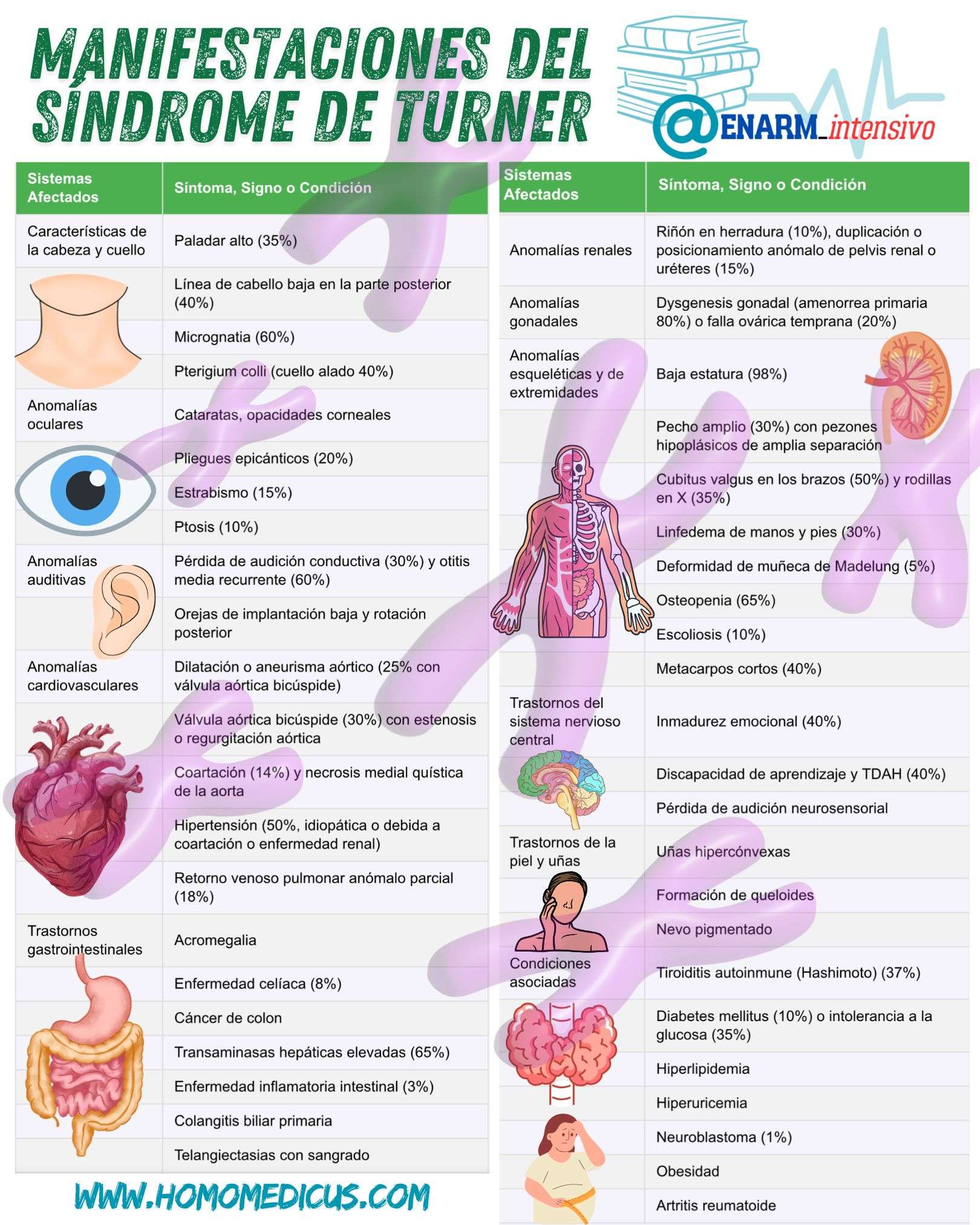
Entre las alteraciones más frecuentes en el área de la cabeza y el cuello, se incluyen un paladar ojival, presente en aproximadamente el 35% de las pacientes, que es indicativo de un desarrollo anómalo del techo del paladar. El línea de cabello posterior baja afecta al 40% de las personas con Turner, lo que puede conferir una apariencia característica en el cuello. La micrognatia, o mandíbula anormalmente pequeña, está presente en un 60% de los casos, contribuyendo a una forma facial distintiva. Además, un cuello alado (pterigium colli) es una manifestación común en un 40% de las pacientes y se refiere a la presencia de pliegues de piel adicionales a ambos lados del cuello, lo que también contribuye a una apariencia física característica.
Anomalías oculares
Las cataratas y las opacidades corneales son comunes en el síndrome de Turner, lo que puede afectar la visión y aumentar el riesgo de otras afecciones oculares. Además, el pliegue epicántico se presenta en el 20% de los casos, una característica relacionada con alteraciones en el pliegue de la piel en la zona interna de los ojos. El estrabismo (15%) y la ptosis palpebral (10%) son alteraciones adicionales en la motilidad ocular y el control de los párpados, que pueden contribuir a la disfunción visual.
Anomalías en los oídos
El pérdida auditiva conductiva es una de las manifestaciones más comunes, afectando aproximadamente al 30% de las pacientes. Esta condición está generalmente asociada con la presencia de otitis media recurrente, que afecta al 60% de las personas con Turner y está relacionada con la disfunción del oído medio. Además, las orejas de implantación baja y rotadas hacia atrás son típicas en el 40% de los casos, lo que también contribuye al fenotipo distintivo del síndrome.
Anomalías cardiovasculares
Las anomalías cardiovasculares son una de las complicaciones más graves y comunes en el síndrome de Turner. Se observa una dilatación o aneurisma de la aorta en el 25% de los casos, a menudo asociada con la presencia de una válvula aórtica bicúspide en un 30% de las pacientes, lo que puede provocar estenosis o regurgitación aórtica. La coartación de la aorta, que afecta al 14% de las pacientes, es otra manifestación importante que puede llevar a hipertensión y otras complicaciones graves si no se trata adecuadamente. Además, aproximadamente un 18% de las pacientes presentan un retorno venoso pulmonar anómalo parcial, lo que puede contribuir a alteraciones en la circulación pulmonar.
Trastornos gastrointestinales
En el ámbito gastrointestinal, el síndrome de Turner se asocia con una mayor predisposición a ciertas afecciones como la aclorhidria, que es una reducción en la producción de ácido gástrico, y la enfermedad celíaca, que afecta aproximadamente al 8% de las pacientes. Las transaminasas hepáticas elevadas se encuentran en el 65% de las pacientes, lo que sugiere un compromiso hepático, y algunas mujeres también presentan enfermedad inflamatoria intestinal (3%) o colangiopatía biliar primaria. Además, las telangiectasias con tendencia al sangrado son comunes en la piel, lo que puede contribuir a problemas de sangrado inexplicables.
Anomalías renales
El 60% de las pacientes con síndrome de Turner presentan anomalías renales, siendo las más comunes el riñón en herradura (10%), y la duplicación o posición anómala de los uréteres o la pelvis renal (15%). Estas anomalías pueden predisponer a las pacientes a infecciones urinarias recurrentes y a otros problemas renales.
Anomalías gonadales
La disgenesia gonadal es una de las manifestaciones centrales del síndrome de Turner, y se traduce principalmente en amenorrea primaria en el 80% de las pacientes, o en un fallo ovárico precoz en el 20% restante. Estas alteraciones conducen a la infertilidad, que es una característica cardinal de la enfermedad, aunque algunas mujeres con mosaicismo pueden experimentar una función ovárica parcial.
Anomalías esqueléticas y de extremidades
Las alteraciones en el sistema esquelético son comunes en el síndrome de Turner. La baja estatura es una de las manifestaciones más destacadas y está presente en casi todas las pacientes (98%). Otras anomalías incluyen un tórax en escudo ancho (30%), asociado a pezones hipoplásicos y ampliamente espaciados. Un 50% de las pacientes presentan cúbito valgo (desviación hacia fuera de los codos), y un 35% presentan rodillas en valgo. La linfedema de las manos y los pies se observa en aproximadamente el 30% de las pacientes, lo que contribuye a la apariencia hinchada de las extremidades en la infancia. Además, algunas pacientes desarrollan deformidad de Madelung en las muñecas (5%) y osteopenia (65%), que puede predisponer a fracturas óseas. La escoliosis también se presenta en un 10% de las pacientes, y los cuartos metacarpos cortos (40%) son una característica esquelética distintiva.
Trastornos del sistema nervioso central
Las dificultades emocionales y cognitivas son comunes en el síndrome de Turner. Aproximadamente el 40% de las pacientes presentan inmadurez emocional, lo que puede manifestarse como dificultades en la regulación emocional y social. Además, las dificultades de aprendizaje y el trastorno por déficit de atención e hiperactividad (TDAH) son frecuentes en el 40% de las pacientes, lo que puede afectar el rendimiento académico y la adaptación escolar. Las pérdidas de audición sensorineural también son comunes.
Trastornos de la piel y las uñas
Las pacientes con síndrome de Turner tienen mayor probabilidad de desarrollar uñas hipercónvexas, formación de queloides y nevos pigmentados, lo que indica una predisposición a trastornos dérmicos y a cambios en la pigmentación de la piel.
Condiciones asociadas
Además de las manifestaciones clínicas directas del síndrome de Turner, las pacientes tienen una mayor predisposición a diversas condiciones asociadas, como la tiroiditis autoinmune de Hashimoto (37%), la diabetes mellitus o intolerancia a la glucosa (35%), hiperlipidemia, hiperuricemia, y en menor medida, neuroblastoma (1%). También es frecuente la obesidad, y algunas pacientes pueden desarrollar artritis reumatoide.
Exámenes complementarios
Hallazgos de laboratorio
Uno de los principales componentes del diagnóstico del síndrome de Turner es la identificación de hipogonadismo, que se confirma mediante el análisis de los niveles séricos de hormonas gonadotrópicas. Las pacientes con síndrome de Turner presentan una falta de desarrollo adecuado de las gónadas, lo que conduce a niveles elevados de las hormonas hormona foliculoestimulante (FSH) y hormona luteinizante (LH). Estas hormonas, que normalmente se elevan en la pubertad como parte del proceso de maduración sexual, se encuentran anormalmente altas en niñas afectadas por el síndrome de Turner debido a la disfunción gonadal primaria. La falta de una respuesta adecuada por parte de los ovarios, debido a su disgenesia o ausencia, provoca la liberación desproporcionada de FSH y LH en un intento del cuerpo por estimular los ovarios.
Para confirmar el diagnóstico genético del síndrome de Turner, se realiza un cariotipo sanguíneo, que revela la presencia de un cariotipo 45,XO, lo que significa que hay una ausencia de un cromosoma X en las células somáticas, o bien, se detectan otras anomalías cromosómicas relacionadas, como mosaico con variaciones en la constitución cromosómica (por ejemplo, 45,XO/46,XX). En algunos casos, también pueden observarse anomalías del cromosoma X, como cromosomas en anillo o deleciones parciales, lo que sugiere una forma menos clásica del síndrome de Turner. El diagnóstico definitivo depende del análisis detallado del cariotipo, que permite identificar las características genéticas particulares del trastorno.
En cuanto a los niveles de hormona del crecimiento (GH) y factor de crecimiento similar a la insulina tipo 1 (IGF-1), estos suelen encontrarse dentro de los rangos normales. A pesar de la baja estatura característica del síndrome de Turner, los niveles de estas hormonas no presentan alteraciones significativas. Esto es relevante porque, aunque las pacientes con síndrome de Turner pueden tener una estatura final significativamente baja, la disfunción en su crecimiento no se debe a una deficiencia de hormona del crecimiento, sino más bien a la falta de otros factores relacionados con el desarrollo hormonal y físico.
Imágenes
El diagnóstico del síndrome de Turner no se limita a las pruebas de laboratorio, sino que también involucra estudios de imagen detallados para evaluar posibles anomalías cardíacas, aórticas y renales, las cuales son comunes en este síndrome. Dado que las alteraciones cardiovasculares y renales son complicaciones importantes del síndrome de Turner, es crucial realizar un ultrasonido transtorácico y una resonancia magnética (RM) de tórax y abdomen en todas las pacientes para identificar la presencia de estas anomalías y planificar un manejo adecuado.
El ultrasonido transtorácico permite visualizar estructuras cardíacas y aórticas, facilitando la detección de condiciones como la coartación de la aorta, la válvula aórtica bicúspide, la dilatación o aneurisma aórtico, y otros defectos cardiovasculares que pueden requerir intervención quirúrgica o seguimiento a largo plazo. Además, el ultrasonido es útil para evaluar la función y la morfología de los riñones, dado que las anomalías renales, como el riñón en herradura y la duplicación de los uréteres, son relativamente frecuentes en las pacientes con síndrome de Turner.
La resonancia magnética complementa estos estudios al proporcionar una visión más detallada de las estructuras internas, especialmente de la aorta y otras áreas críticas, permitiendo una evaluación más precisa de las condiciones potencialmente graves que pueden implicar un riesgo para la salud a largo plazo. La evaluación mediante RM también es importante para la detección de anomalías que no sean evidentes en los estudios de imagen convencionales, como ciertas malformaciones renales o estructuras vasculares complejas.
Tratamiento
El tratamiento para la baja estatura en el síndrome de Turner se centra principalmente en la utilización de terapias hormonales que promuevan un crecimiento adecuado durante los primeros años de vida y la adolescencia. La intervención temprana es crucial para maximizar el potencial de crecimiento y minimizar los efectos de la disgenesia gonadal y las deficiencias hormonales típicas de esta condición.
La terapia con hormona del crecimiento (GH) es uno de los pilares del tratamiento para la baja estatura en pacientes con síndrome de Turner. Este tratamiento debe iniciarse lo más temprano posible, idealmente entre los 4 y los 6 años de edad, y antes de los 12 años, para optimizar los resultados y aprovechar el período de mayor plasticidad del crecimiento óseo. El tratamiento con GH tiene como objetivo incrementar la velocidad de crecimiento, ayudando a las pacientes a alcanzar una estatura más cercana a la media para su género y edad.
La dosis estándar de hormona de crecimiento es de 50 microgramos por kilogramo por día o 4.5 unidades internacionales por metro cuadrado de superficie corporal por día. Esta dosis se ajusta de manera individualizada según los niveles séricos de factor de crecimiento similar a la insulina tipo 1 (IGF-I), que debe mantenerse dentro de un rango de 3 desviaciones estándar por encima de la media para la edad. La monitorización de los niveles de IGF-I es crucial para ajustar la dosis de GH de manera que se logre un efecto terapéutico óptimo sin generar efectos secundarios.
Aunque los efectos secundarios de la GH son raros, uno de los más infrecuentes, pero graves, es la presencia de pseudotumor cerebri, una condición en la que se presenta una presión intracraneal elevada sin una masa cerebral evidente. Esta complicación requiere vigilancia clínica, especialmente al inicio del tratamiento.
En casos en los que el tratamiento con GH no sea suficiente para promover un crecimiento adecuado, se agrega un andrógeno oral, como el oxandrolona, después de los 10 años de edad. Este medicamento, que se administra en dosis de 0.03 a 0.05 miligramos por kilogramo por día, tiene la capacidad de estimular el crecimiento óseo y mejorar el aumento de altura en las niñas con síndrome de Turner que no responden de manera adecuada solo a la GH. La oxandrolona funciona al mimetizar parcialmente los efectos de los andrógenos, promoviendo el crecimiento en el contexto de un desarrollo puberal incompleto o ausente, típico de este síndrome.
La terapia hormonal en el síndrome de Turner también incluye la administración de estrógenos y, eventualmente, progesterona para inducir el desarrollo sexual secundario y simular el inicio de la pubertad. Después de los 12 años, se inicia un tratamiento con estradiol transdérmico en dosis bajas, con una aumento gradual de la dosis durante un período de 2 a 3 años. Este enfoque busca inducir cambios físicos típicos de la pubertad, como el desarrollo mamario, la distribución corporal de la grasa y la maduración del aparato reproductor. La gradualidad en el aumento de la dosis es importante para reducir los riesgos de efectos adversos y asegurar que el desarrollo puberal siga un curso fisiológico y controlado.
Tras dos años de tratamiento con estrógenos, o si se presenta sangrado menstrual, se inicia el tratamiento con progesterona, lo que permite la inducción de un ciclo menstrual regular. La progesterona se administra para provocar la descarte de endometrial, lo que también es crucial para prevenir la hiperplasia endometrial, que puede ocurrir debido a la exposición prolongada a estrógenos sin la contraposición de progesterona.
Es fundamental que el tratamiento de las pacientes con síndrome de Turner sea multidisciplinario, ya que involucra no solo el manejo de la baja estatura, sino también la atención a las posibles complicaciones cardiovasculares, renales, emocionales y reproductivas. El tratamiento con GH, andrógenos, y estrógenos debe ser supervisado de cerca por endocrinólogos pediátricos, ginecólogos y otros especialistas, que ajusten las dosis según las necesidades individuales de cada paciente.
El seguimiento regular de los efectos secundarios y la monitorización de la respuesta al tratamiento es crucial. Esto incluye la evaluación continua de los niveles hormonales, la vigilancia de los parámetros de crecimiento, y el monitoreo de las complicaciones asociadas, como las afecciones cardiovasculares o renales.
Complicaciones y Vigilancia
Las mujeres con síndrome de Turner presentan un riesgo significativo de diversas complicaciones a lo largo de su vida, lo que contribuye a una esperanza de vida reducida en comparación con la población general. Estos riesgos se deben a un conjunto de factores relacionados con la disfunción hormonal, la predisposición genética y las malformaciones asociadas a este síndrome, que afectan principalmente a sistemas cardiovasculares, metabólicos, óseos y renales.
Entre las complicaciones más comunes que enfrentan las mujeres con síndrome de Turner se encuentran la diabetes mellitus (tipos 1 y 2), la hipertensión, la dislipidemia y la osteoporosis. Estas condiciones metabólicas y cardiovasculares contribuyen significativamente al riesgo de morbilidad y mortalidad prematura. La diabetes mellitus, por ejemplo, se presenta con una prevalencia elevada, especialmente tipo 2, debido a factores como la resistencia a la insulina, que es común en estas pacientes. La hipertensión y la dislipidemia, por otro lado, están asociadas tanto con alteraciones en la función endotelial como con defectos estructurales en los vasos sanguíneos, lo que incrementa el riesgo de enfermedades cardiovasculares, incluyendo infartos de miocardio y accidentes cerebrovasculares.
La osteoporosis también es una complicación importante, relacionada con la deficiencia de estrógenos, que no solo afecta el crecimiento óseo durante la adolescencia, sino que también pone a las pacientes en un riesgo aumentado de fracturas y pérdida de densidad ósea a lo largo de su vida. La baja estatura y los problemas de alineación esquelética pueden complicar aún más la condición ósea, haciendo que la monitorización de la densidad mineral ósea sea esencial.
Para mitigar estos riesgos, se recomienda que las mujeres con síndrome de Turner realicen un seguimiento anual, que incluya la determinación de la presión arterial y evaluaciones de laboratorio completas. Las pruebas de laboratorio deben incluir la medición de los niveles de hormona estimulante de la tiroides (TSH), enzimas hepáticas, nitrógeno ureico en sangre (BUN), creatinina, y lípidos séricos en ayuno, así como la medición de glucosa en ayuno. Estas pruebas son fundamentales para detectar precozmente cualquier alteración metabólica, hepática o renal que pueda surgir, permitiendo una intervención temprana.
Además, las pruebas auditivas (audiometrías) son recomendadas de manera periódica, cada 1 a 5 años, debido a la alta prevalencia de pérdida auditiva conductiva y otras alteraciones del oído medio, como la otitis media recurrente. La pérdida auditiva puede empeorar con el tiempo si no se detecta y maneja adecuadamente.
La evaluación de la densidad mineral ósea mediante densitometrías óseas debe realizarse de forma periódica, especialmente en mujeres mayores de 18 años, debido a su predisposición a la osteoporosis. Este control es esencial para prevenir fracturas y otras complicaciones óseas, garantizando la intervención temprana con tratamiento hormonal o farmacológico adecuado, si es necesario, para fortalecer los huesos y prevenir su deterioro.
Las anomalías cardiovasculares son una de las principales preocupaciones en las pacientes con síndrome de Turner. Las válvulas aórticas bicúspides son comunes en estas pacientes, y se asocian con un riesgo elevado de varias complicaciones graves, como endocarditis infecciosa, estenosis o regurgitación valvular aórtica, y dilatación de la raíz de la aorta ascendente, que puede evolucionar a una disección aórtica. El riesgo de disección aórtica es más de 100 veces mayor en mujeres con síndrome de Turner que en la población general, lo que resalta la importancia de una vigilancia cardiovascular rigurosa. La conexión venosa pulmonar anómala parcial, que puede llevar a un desvío de izquierda a derecha de la sangre, también es una condición asociada con el síndrome de Turner y puede contribuir a la sobrecarga del corazón derecho y a complicaciones pulmonares si no se detecta y trata a tiempo.
En cuanto a los problemas renales, las mujeres con síndrome de Turner tienen un riesgo elevado de anomalías estructurales renales. Aquellas con el cariotipo clásico 45,XO presentan una mayor prevalencia de malformaciones renales, mientras que aquellas con mosaico 46,X/abnormal X son más propensas a sufrir malformaciones del sistema urinario, particularmente en lo que respecta al sistema de recolección urinaria. Estas malformaciones pueden incluir riñones en herradura, duplicación de uréteres o malformaciones de la pelvis renal, lo que puede afectar la función renal y aumentar el riesgo de infecciones urinarias recurrentes y otras complicaciones renales. La vigilancia mediante estudios de imagen, como ecografías renales, es esencial para detectar y gestionar estas anomalías.
En el síndrome de Turner, las anomalías electrocardiográficas también son frecuentes, particularmente el síndrome de QT largo, que aumenta el riesgo de arritmias y puede poner en peligro la vida de las pacientes. Es importante realizar electrocardiogramas (ECG) de forma regular para monitorizar cualquier alteración en la conducción cardíaca y tomar medidas preventivas, como la modificación de los factores de riesgo y, si es necesario, el tratamiento farmacológico.
Variantes del síndrome de Turner
1. Cariotipo 46,X (Cromosoma X Anómalo)
En algunos casos de síndrome de Turner, la alteración cromosómica implica una deleción pequeña en el brazo corto distal del cromosoma X (denominado Xp-), que incluye el gen SHOX (Short Stature Homeobox Gene). Este gen está relacionado con el crecimiento óseo y la estatura, por lo que su deleción suele provocar baja estatura y anomalías esqueléticas. Las mujeres afectadas por esta variante del síndrome de Turner suelen presentar características clínicas más suaves en comparación con la forma clásica del trastorno.
Aunque la baja estatura y las anomalías óseas son comunes, una de las características distintivas de esta variante es que las pacientes suelen tener un bajo riesgo de insuficiencia ovárica. Es decir, aunque pueden experimentar alguna forma de disfunción gonadal, no es tan frecuente que presenten insuficiencia ovárica primaria o que desarrollen problemas graves de fertilidad, como ocurre en el síndrome de Turner clásico. Esta menor afectación gonadal es un aspecto importante, ya que permite que algunas pacientes con esta variante mantengan una función ovárica relativamente conservada, lo que podría influir en su fertilidad y en el manejo de su desarrollo sexual.
2. Mosaicismo 45,XO/46,XX y 45,XO/46,XY
El mosaicismo es otra variante genética que se observa en el síndrome de Turner y que implica la presencia de dos líneas celulares diferentes en el mismo individuo. Un ejemplo común es el mosaicismo 45,XO/46,XX, en el que algunas células tienen el karyotipo clásico de Turner (45,XO), mientras que otras tienen un karyotipo normal femenino (46,XX). Este tipo de mosaicismo resulta en una forma modificada del síndrome de Turner, que presenta algunas diferencias significativas en cuanto a las manifestaciones clínicas.
Las pacientes con este tipo de mosaicismo tienden a ser más altas que aquellas con el karyotipo clásico 45,XO, lo que sugiere que la presencia de algunas células con un cromosoma X completo tiene un impacto positivo en el crecimiento. Además, este tipo de mosaicismo suele estar asociado con una mejor función gonadal, lo que significa que las pacientes pueden experimentar un desarrollo puberal más completo y tener un menor riesgo de insuficiencia ovárica. Algunas mujeres con este mosaicismo pueden incluso presentar menstruaciones regulares y desarrollar características sexuales secundarias, lo que es menos común en las mujeres con la forma clásica del síndrome de Turner.
En cuanto a otras manifestaciones, las pacientes con mosaicismo 45,XO/46,XX suelen presentar menos características del síndrome de Turner, como malformaciones cardíacas, renales y esqueléticas. Este patrón de presentación más leve hace que el diagnóstico y manejo del síndrome sea más complejo, ya que algunas de las manifestaciones clásicas pueden estar ausentes o ser más sutiles.
En un subgrupo de mujeres con mosaicismo 45,XO/46,XY, la presencia de células con un cromosoma Y puede resultar en una fenotipo ambigua en términos de desarrollo sexual. Estas pacientes pueden presentar genitales ambiguos, lo que podría dificultar la identificación del sexo cromosómico y una evaluación diagnóstica más detallada. Además, estas mujeres pueden experimentar infertilidad masculina, dado que las células con el cromosoma Y pueden interferir con el desarrollo normal de los ovarios y las gónadas. Sin embargo, el fenotipo general de estas pacientes puede ser casi normal en términos de características físicas y desarrollo puberal.
El diagnóstico de estas variantes del síndrome de Turner requiere un análisis genético detallado, específicamente a través de un estudio de cariotipo o técnicas de hibridación fluorescente in situ (FISH), que permiten identificar las anomalías cromosómicas en las células. Estos estudios son cruciales para confirmar el tipo de variante presente y para planificar un manejo adecuado, ya que la naturaleza del tratamiento puede variar según la forma del síndrome.
El manejo de las pacientes con variantes de Turner debe ser personalizado y adaptado a las manifestaciones clínicas específicas de cada variante. En el caso de los pacientes con mosaicismo 45,XO/46,XX, el tratamiento hormonal puede ser menos agresivo, ya que muchas pacientes pueden experimentar una pubertad más natural y tener menos problemas de fertilidad. Por otro lado, las pacientes con la variante de deleción en el cromosoma Xp- pueden requerir un enfoque más centrado en el manejo de la estatura y las anomalías esqueléticas.

Fuente y lecturas recomendadas
- Steiner M et al. Turner syndrome: an update. Adv Pediatr. 2022;69:177. [PMID: 35985709]
- Yoon SH et al. Organ abnormalities caused by Turner syndrome. Cells. 2023;12:1365. [PMID: 37408200]
Síguenos en X: @el_homomedicus y @enarm_intensivo Síguenos en instagram: homomedicus y en Treads.net como: Homomedicus
Aprende administración paso a paso

ADMINISTRACION DESDE CERO
Originally posted on 30 de abril de 2023 @ 10:27 AM