
La enfermedad de Wilson, también conocida como degeneración hepatolenticular, es un trastorno genético autosómico recesivo caracterizado por la acumulación tóxica de cobre en diferentes tejidos del organismo, principalmente en el hígado y el sistema nervioso central. Este padecimiento se origina debido a mutaciones en el gen ATP7B, localizado en el cromosoma 13, que codifica para una proteína transportadora de cobre esencial en la regulación de los niveles de este elemento en el organismo.
En condiciones normales, el cobre es absorbido en el intestino delgado, transportado al hígado y utilizado en procesos metabólicos críticos, como la síntesis de enzimas dependientes de cobre. El exceso de este metal se excreta principalmente a través de la bilis. Sin embargo, en la enfermedad de Wilson, las mutaciones en el gen ATP7B impiden la correcta función de la proteína transportadora, alterando el transporte de cobre hacia la bilis y su incorporación a la ceruloplasmina, una proteína encargada de su movilización en el plasma. Como resultado, el cobre se acumula de manera progresiva en el hígado, lo que lleva a estrés oxidativo, disfunción mitocondrial y eventual daño hepático.
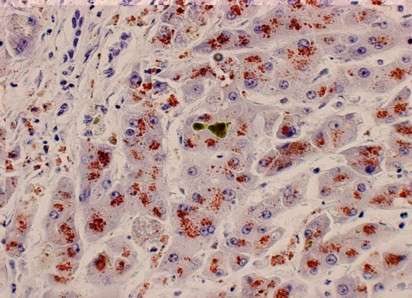
El proceso patológico comienza en el hígado, donde la acumulación inicial de cobre causa inflamación, fibrosis y, en casos avanzados, cirrosis. A medida que se saturan las reservas hepáticas, el cobre libre se libera al torrente sanguíneo, depositándose en otros tejidos, particularmente en el cerebro (donde afecta principalmente a los ganglios basales) y en la córnea (manifestándose como los anillos de Kayser-Fleischer, una de las características clínicas distintivas). Otros órganos, como los riñones y el corazón, también pueden verse afectados, lo que explica la amplia variedad de síntomas sistémicos observados en esta enfermedad.
Desde una perspectiva genética, la mayoría de los pacientes afectados son heterocigotos compuestos, es decir, portadores de dos variantes patogénicas diferentes en el gen ATP7B. Hasta la fecha, se han identificado más de 600 mutaciones en este gen, de las cuales algunas son más prevalentes en poblaciones específicas. Por ejemplo, la mutación H1069Q es común en personas de ascendencia europea del norte, representando hasta el 63 % de los alelos patogénicos en esta población. Estas diferencias en la distribución de las mutaciones contribuyen a variaciones en la presentación clínica y en la gravedad de la enfermedad entre diferentes grupos étnicos.
Manifestaciones clínicas
En los adolescentes, la enfermedad de Wilson suele manifestarse predominantemente como enfermedad hepática. Esta presentación es más común en mujeres jóvenes y puede incluir síntomas como hepatitis crónica, esplenomegalia, hipertensión portal y, en casos severos, insuficiencia hepática aguda. Por otro lado, en los adultos jóvenes, especialmente en hombres, la enfermedad tiende a manifestarse como un trastorno neuropsiquiátrico. Esto puede reflejar diferencias en la sensibilidad de los tejidos al cobre acumulado o variaciones hormonales y metabólicas que influyen en la progresión y expresión de la enfermedad.
Aunque tradicionalmente se ha considerado que los síntomas suelen comenzar antes de los 40 años, cada vez hay más evidencia de que el inicio tardío es más común de lo que se pensaba. Esto subraya la importancia de considerar la enfermedad de Wilson como un posible diagnóstico en personas de todas las edades, especialmente si presentan síntomas compatibles.
El diagnóstico debe ser contemplado en una variedad de contextos clínicos, incluyendo:
- Niños o adultos jóvenes con hepatitis aguda o crónica, esplenomegalia con hiperesplenismo, anemia hemolítica Coombs negativa, hipertensión portal o anomalías neurológicas y psiquiátricas.
- Adultos menores de 40 años con insuficiencia hepática aguda o hepatitis crónica.
- Pacientes con enfermedad hepática aguda grave, donde un índice diagnóstico útil es una relación fosfatasa alcalina (U/L) a bilirrubina total (mg/dL) menor de 4 y una relación AST-ALT superior a 2,2.
El daño hepático en la enfermedad de Wilson puede oscilar desde alteraciones leves en las pruebas de función hepática hasta cirrosis avanzada e insuficiencia hepática. En pacientes con insuficiencia hepática aguda, más frecuente en mujeres, se observan características bioquímicas distintivas, como niveles inusualmente bajos de fosfatasa alcalina, que pueden ser un marcador diferencial importante.
La hipertensión portal secundaria a la cirrosis puede complicar el cuadro clínico, con hallazgos como esplenomegalia e hiperesplenismo, que a menudo se asocian con anemia y trombocitopenia. Estos hallazgos, junto con la anemia hemolítica Coombs negativa, refuerzan la sospecha diagnóstica.
Los síntomas neurológicos resultan de la acumulación de cobre en los ganglios basales, estructuras cerebrales críticas para la coordinación motora y la regulación de movimientos. Entre las manifestaciones más comunes se encuentran:
- Síndrome rígido-acinético, similar al parkinsonismo.
- Pseudosclerosis, caracterizada por temblor.
- Ataxia y síndrome distónico, que afectan el control postural y generan movimientos anormales.
- Otros síntomas incluyen disartria, disfagia, incoordinación, espasticidad, migrañas, insomnio y, en algunos casos, convulsiones.
Las alteraciones psiquiátricas, como cambios en la personalidad, labilidad emocional y riesgo aumentado de depresión, suelen preceder a las manifestaciones neurológicas más características. Estos síntomas pueden ser subestimados o atribuidos erróneamente a otros trastornos, retrasando el diagnóstico.
El signo clínico patognomónico de la enfermedad es el anillo de Kayser-Fleischer, un depósito de cobre en la membrana de Descemet de la córnea. Este anillo, de color marrón o verde grisáceo, es más evidente en los polos superior e inferior de la córnea y, aunque en ocasiones es visible a simple vista, su detección precisa requiere un examen con lámpara de hendidura. Es más frecuente en pacientes con manifestaciones neuropsiquiátricas y puede estar ausente en aquellos con afectación exclusivamente hepática.
La acumulación de cobre también afecta otros sistemas, generando manifestaciones como:
- Alteraciones renales: cálculos, aminoaciduria, acidosis tubular renal.
- Endocrinopatías: hipoparatiroidismo, infertilidad.
- Manifestaciones hematológicas: anemia hemolítica.
- Lesiones cutáneas: lipomas subcutáneos.
La variabilidad clínica de la enfermedad de Wilson, combinada con su baja prevalencia, puede llevar a un diagnóstico tardío. Sin embargo, su identificación precoz es esencial, ya que el tratamiento oportuno con agentes quelantes de cobre (como penicilamina o trientina) o compuestos de zinc puede prevenir complicaciones graves y mejorar significativamente la calidad de vida. Además, el trasplante hepático puede ser una opción para pacientes con insuficiencia hepática terminal.
Exámenes diagnósticos.
El diagnóstico de la enfermedad de Wilson es particularmente desafiante debido a la amplia variabilidad clínica de sus manifestaciones, la superposición con otros trastornos hepáticos o neurológicos y las limitaciones de las herramientas diagnósticas existentes. Aunque se han desarrollado sistemas de puntuación como los criterios de Leipzig y el Índice de Wilson para facilitar la evaluación, estos no siempre son suficientes para establecer un diagnóstico definitivo, lo que subraya la necesidad de un enfoque integral y multimodal.
Marcadores bioquímicos clave
El diagnóstico de la enfermedad de Wilson se basa principalmente en la detección de alteraciones en la homeostasis del cobre. Entre los hallazgos más relevantes se incluyen:
- Excreción urinaria de cobre aumentada:
- En individuos afectados, la excreción de cobre en orina suele superar los 40 microgramos por 24 horas, y con frecuencia excede los 100 microgramos por 24 horas. Este hallazgo, aunque útil, no es exclusivo de la enfermedad de Wilson, ya que también puede observarse en otras condiciones que alteran la función hepática o renal.
- Ceruloplasmina sérica reducida:
- Los niveles bajos de ceruloplasmina (menos de 14 miligramos por decilitro) son una característica común. Valores inferiores a 10 miligramos por decilitro refuerzan fuertemente la sospecha diagnóstica. Sin embargo, la ceruloplasmina puede estar en el rango normal en algunos pacientes, especialmente aquellos con formas tardías o atípicas, y puede estar disminuida en otras condiciones, como enfermedades hepáticas avanzadas o deficiencia nutricional.
- Concentración elevada de cobre hepático:
- La acumulación de cobre en el tejido hepático es un hallazgo característico. Una concentración superior a 250 microgramos por gramo de tejido hepático seco se considera diagnóstica. Sin embargo, la distribución desigual del cobre en el hígado puede limitar la sensibilidad de este criterio, especialmente en etapas iniciales de la enfermedad.
- Relación cobre intercambiable-serocobre total:
- Este indicador, también conocido como “cobre intercambiable relativo”, mide la fracción de cobre no unido a la ceruloplasmina en el plasma. Un valor superior al 18,5 % se ha identificado como una prueba altamente confiable y específica para el diagnóstico de la enfermedad de Wilson, superando en sensibilidad a la excreción urinaria de cobre.
- Ensayo enzimático de ceruloplasmina:
- Este método mide la actividad enzimática de la ceruloplasmina y se considera más sensible que los niveles séricos de ceruloplasmina determinados por inmunoensayo. Puede ser particularmente útil en casos ambiguos o atípicos.
Evaluaciones clínicas y de imagen
- Anillos de Kayser-Fleischer:
- Estos depósitos de cobre en la membrana de Descemet de la córnea son un hallazgo patognomónico, pero no siempre están presentes. Su ausencia es común en pacientes con manifestaciones exclusivamente hepáticas, mientras que su presencia es casi universal en aquellos con síntomas neurológicos.
- Biopsia hepática:
- Además de confirmar la acumulación de cobre, la biopsia hepática puede revelar características histológicas asociadas, como hepatitis aguda o crónica, esteatosis o cirrosis. Este procedimiento sigue siendo una herramienta crucial cuando las pruebas no invasivas no son concluyentes.
- Resonancia magnética cerebral:
- En pacientes con manifestaciones neurológicas, la resonancia magnética puede mostrar depósitos de cobre en los ganglios basales, el tallo cerebral y el cerebelo, incluso en etapas tempranas. Estos hallazgos pueden correlacionarse con síntomas específicos, como rigidez, temblores y ataxia.
Pruebas genéticas
El análisis molecular del gen ATP7B representa el estándar de oro para confirmar el diagnóstico en casos dudosos. La identificación de dos variantes patogénicas en este gen (en estado homocigoto o heterocigoto compuesto) es diagnóstica de la enfermedad. Dado que más de 600 mutaciones han sido descritas, la secuenciación completa del gen es a menudo necesaria, especialmente en poblaciones donde las variantes comunes son menos prevalentes. Este enfoque también permite el diagnóstico precoz en familiares asintomáticos de pacientes afectados.
Limitaciones diagnósticas
Pese a los avances en las herramientas diagnósticas, estas tienen limitaciones significativas:
- Falsos negativos: Los niveles de ceruloplasmina pueden ser normales en algunas personas afectadas, especialmente aquellas en etapas tempranas o con formas atípicas.
- Falsos positivos: La excreción urinaria de cobre y los niveles de ceruloplasmina pueden alterarse en otras enfermedades hepáticas, lo que reduce la especificidad de estas pruebas.
- Distribución desigual del cobre hepático: Esto puede llevar a subestimaciones en la biopsia hepática, especialmente si la muestra no es representativa.
- Variabilidad en las manifestaciones clínicas: Los síntomas pueden ser tan diversos que el diagnóstico no siempre es considerado inicialmente, lo que retrasa el tratamiento.
Tratamiento
El tratamiento temprano de la enfermedad de Wilson es fundamental para prevenir el daño progresivo causado por la acumulación tóxica de cobre en el hígado, el sistema nervioso central y otros tejidos. Este enfoque terapéutico requiere una combinación de restricciones dietéticas, farmacoterapia específica y, en casos avanzados, intervenciones más agresivas como el trasplante hepático. Además, la detección oportuna de la enfermedad en familiares de primer grado es clave para prevenir complicaciones graves en portadores asintomáticos.
El cobre acumulado en los tejidos actúa como un agente tóxico que desencadena estrés oxidativo, inflamación y daño celular. En el hígado, esto puede conducir a hepatitis, fibrosis, cirrosis e insuficiencia hepática. En el sistema nervioso central, los depósitos de cobre afectan estructuras como los ganglios basales, el tallo cerebral y el cerebelo, generando síntomas neurológicos y psiquiátricos irreversibles si no se aborda a tiempo. Por lo tanto, la eliminación temprana del exceso de cobre y la prevención de su acumulación futura son esenciales para preservar la función hepática y neurológica.
Restricción dietética
Aunque útil como complemento del tratamiento farmacológico, la restricción dietética por sí sola no es suficiente para controlar la enfermedad. Los alimentos ricos en cobre, como mariscos, vísceras, frutos secos, champiñones, productos de soya y chocolate, deben evitarse para minimizar la ingesta exógena. Sin embargo, dado que la acumulación de cobre en la enfermedad de Wilson está principalmente relacionada con un defecto en su excreción hepática, se requiere farmacoterapia para lograr una eliminación efectiva.
Terapia farmacológica
D-penicilamina
La D-penicilamina ha sido el tratamiento de referencia durante décadas. Este agente quelante actúa al unirse al cobre y aumentar su excreción urinaria. La dosis habitual varía entre 0,75 y 2 gramos al día, administrada en dosis divididas. No obstante, su uso está limitado por efectos adversos significativos, que incluyen:
- Reacciones alérgicas.
- Supresión de la médula ósea.
- Proteinuria y nefrotoxicidad.
- Síntomas gastrointestinales.
Debido a estas limitaciones, muchos pacientes no toleran la D-penicilamina, lo que lleva a la búsqueda de alternativas terapéuticas.
Trientina
La trientina es otro agente quelante eficaz con un perfil de seguridad más favorable. Se administra en dosis de 250 a 500 miligramos tres veces al día y es particularmente útil en pacientes que no toleran la D-penicilamina. En 2022, se aprobó una nueva formulación, trientina tetrahidrocloruro, diseñada específicamente para el mantenimiento a largo plazo, con una menor incidencia de efectos adversos gastrointestinales y una mayor adherencia al tratamiento.
Zinc
El zinc, administrado como acetato o gluconato en dosis de 50 miligramos tres veces al día, tiene un mecanismo de acción diferente al de los agentes quelantes. Este elemento interfiere con la absorción intestinal de cobre al inducir la producción de metalotioneína en las células intestinales, atrapando el cobre y promoviendo su excreción fecal. Es particularmente útil como terapia de mantenimiento después de la fase inicial de eliminación de cobre o en combinación con agentes quelantes. Además, el zinc es bien tolerado y presenta un perfil de efectos secundarios mínimo, lo que lo convierte en una opción atractiva para el manejo a largo plazo.
Vitamina E
La vitamina E, un antioxidante liposoluble, ha sido recomendada como complemento terapéutico en la enfermedad de Wilson para reducir el estrés oxidativo asociado con la acumulación de cobre. Sin embargo, su efectividad no ha sido rigurosamente estudiada en ensayos clínicos, por lo que su uso se basa más en consideraciones teóricas que en evidencia robusta.
Tratamiento durante el embarazo
El manejo de la enfermedad de Wilson durante el embarazo requiere ajustes cuidadosos en la terapia para minimizar el riesgo tanto para la madre como para el feto. La dosis de D-penicilamina o trientina debe reducirse para evitar efectos adversos potenciales, como el retraso en la cicatrización de heridas o el riesgo de defectos congénitos. El zinc se considera seguro durante el embarazo y puede ser utilizado como terapia de mantenimiento.
Trasplante hepático
En casos de insuficiencia hepática aguda o cirrosis descompensada, el trasplante de hígado es la única opción terapéutica definitiva. Este procedimiento no solo reemplaza el órgano dañado, sino que también corrige el defecto metabólico subyacente al proporcionar un hígado funcional capaz de manejar el metabolismo del cobre de manera normal.
Tamizaje en familiares
Dado que la enfermedad de Wilson es un trastorno autosómico recesivo, los familiares de primer grado de los pacientes tienen un riesgo significativo de ser portadores o de desarrollar la enfermedad. El tamizaje debe incluir:
- Pruebas bioquímicas hepáticas: Para detectar alteraciones hepáticas tempranas.
- Ceruloplasmina sérica: Para identificar niveles bajos sugestivos.
- Examen con lámpara de hendidura: Para evaluar la presencia de anillos de Kayser-Fleischer.
- Análisis genético: Si la variante patogénica en el gen ATP7B ha sido identificada en el paciente índice, se puede realizar una prueba dirigida en los familiares.
El tratamiento de la enfermedad de Wilson es de por vida, y la falta de adherencia puede llevar a la reaparición de los síntomas y a un daño irreversible en los órganos afectados. Es esencial educar a los pacientes sobre la importancia del tratamiento continuo y monitorear regularmente su estado clínico y bioquímico para ajustar la terapia según sea necesario.

Fuente y lecturas recomendadas:
- Alkhouri N et al. Wilson disease: a summary of the updated AASLD Practice Guidance. Hepatol Commun. 2023;7:e0150. [PMID: 37184530]
- Mazhar A et al. Updates on Wilson disease. Clin Liver Dis (Hoboken). 2023;22:117. [PMID: 3790886]
- Roberts EA et al. Current and emerging issues in Wilson’s disease. N Engl J Med. 2023;389:922. [PMID: 37672695]
Aprende administración paso a paso

ADMINISTRACION DESDE CERO
Originally posted on 16 de enero de 2025 @ 7:46 PM

