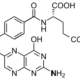
Formas químicas
Hay diversas formas químicas de cobalamina (vitamina B12), pero todas poseen un átomo de cobalto en el centro de un anillo de corrina. En la naturaleza, la vitamina se encuentra sobre todo en la forma de 2-desoxiadenosil (ado), situado en las mitocondrias. Es el cofactor para la enzima metilmalonil-CoA mutasa.
La otra cobalamina natural importante es la metilcobalamina, que es la forma localizada en el plasma humano y el citoplasma celular. Es el cofactor para la metionina sintasa.
Existen cantidades menores de hidroxocobalamina, que es en lo que se convierten con rapidez la metilcobalamina y adocobalamina al exponerse a la luz solar.
Fuentes y requerimientos
La única fuente de cobalamina para el ser humano son los alimentos de origen animal como carne, pescado y productos lácteos. Las verduras, frutas y otros alimentos que no son de origen animal carecen de cobalamina, a menos que estén contaminados con bacterias.
La alimentación occidental regular proporciona entre 5 y 30 µg diarios de cobalamina.
La eliminación diaria de esta vitamina en los adultos, sobre todo en la orina y las heces, fluctúa entre 1 y 3 µg (cerca de 0.1% de las reservas del organismo) y, dado que el organismo no tiene la capacidad para degradar cobalamina, las necesidades diarias también son de alrededor de 1 a 3 µg.
Las reservas corporales son del orden de 2 a 3 mg, suficientes para tres a cuatro años si se interrumpe por completo el aporte.
Absorción de cobalamina por el organismo
Hay dos mecanismos para la absorción de cobalamina. Uno es pasivo, que ocurre por igual a través de las mucosas bucal, duodenal e ileal; es rápido pero en extremo ineficaz y permite absorber <1% de una dosis oral.
El mecanismo fisiológico habitual es activo; se lleva a cabo a través del íleon, es eficaz para dosis orales pequeñas de cobalamina y está mediado por el factor intrínseco gástrico.
La cobalamina de los alimentos se libera de complejos proteínicos por enzimas dispuestas en el estómago, duodeno y yeyuno; se combinan de manera rápida con una glucoproteína salival que pertenece a la familia de las proteínas de unión a cobalamina que se conocen como haptocorrinas.
En el intestino la haptocorrina es digerida por la tripsina pancreática y la cobalamina es transferida al factor intrínseco. El factor intrínseco se produce en las células parietales gástricas del fondo y cuerpo gástricos y su secreción es paralela a la del ácido clorhídrico. En condiciones normales la producción de factor intrinseco es abundante. El complejo factor intrínseco-cobalamina pasa al íleon, donde el factor intrínseco se adhiere a un receptor específico (cubilina) ubicado en la membrana de la microvellosidad de los enterocitos. Al parecer transita por medio de la AMN (amnionless), proteína del receptor endocítico que dirige la ubicación y endocitosis de la cubilina con su ligando complejo factor intrínseco-cobalamina.
El complejo cobalamina-factor intrínseco penetra en la célula ileal donde se destruye el factor intrínseco. Después de un retraso de unas 6 horas, la cobalamina aparece en la sangre de la vena porta unida a la transcobalamina II.
Todos los días entran a la bilis entre 0.5 y 5.0 µg de cobalamina. Su molécula se une al factor intrínseco y normalmente una parte importante de la cobalamina biliar se reabsorbe al mismo tiempo que la porción derivada de las células intestinales esfaceladas. Gracias a la gran cantidad de cobalamina que viaja en la circulación enterohepática, su deficiencia aparece con más rapidez en las personas con una absorción deficiente de cobalamina que en los vegetarianos, en quienes la reabsorción biliar del compuesto se encuentra intacta.
Transporte de cobalamina en sangre
El plasma humano contiene dos proteínas principales que transportan cobalamina; ambas se unen a esta vitamina molécula a molécula. Existe una haptocorrina conocida como transcobalamina I que guarda una relación cercana con otras haptocorrinas de unión a cobalamina que se encuentran en la leche, el jugo gástrico, la bilis, la saliva y otros líquidos.
La transcobalamina I se deriva principalmente de gránulos específicos en los neutrófilos. En general, 66% se encuentra saturado con cobalamina, que se une con fuerza. La transcobalamina I no estimula la penetración de cobalamina en los tejidos. Los receptores de glucoproteínas en las células hepáticas ayudan a extraer transcobalamina I del plasma.
La transcobalamina I es muy importante en el transporte de los análogos de cobalamina hacia el hígado para su excreción hepática en la bilis.
La otra proteína importante para transportar cobalamina en el plasma es transcobalamina II. Es sintetizada por el hígado y otros tejidos, en especial macrófagos, íleon y endotelio vascular. Lo normal es que transporte sólo 20 a 60 ng de cobalamina/L de plasma y la libere con facilidad hacia la médula, placenta y otros tejidos, donde penetra por medio de endocitosis mediada por receptores en la que participan el receptor de transcobalamina II y la megalina. El complejo TC II-cobalamina es fagocitado por endocitosis a través de grietas revestidas con clatrina; el complejo se degrada, pero es probable que el receptor sea reciclado hacia la membrana celular como sucede con la transferrina.
La cobalamina “libre” se exporta a través de un transportador de fármacos tipo casete de unión a ATP llamado también proteína 1 de resistencia a múltiples fármacos.

Aprende administración paso a paso

ADMINISTRACION DESDE CERO