La colangitis biliar primaria es una enfermedad hepática crónica que se caracteriza principalmente por la destrucción autoinmune de los pequeños conductos biliares intrahepáticos, lo que conduce a la colestasis, es decir, a la obstrucción del flujo biliar. Esta condición afecta principalmente a mujeres de mediana edad, generalmente entre los 40 y 60 años, y se desarrolla de manera insidiosa, lo que significa que los síntomas suelen ser sutiles y progresivos, lo que dificulta un diagnóstico temprano. A menudo, la enfermedad se detecta de manera incidental en análisis de sangre, cuando se encuentran niveles elevados de fosfatasa alcalina, un marcador de obstrucción biliar.
La denominación «colangitis biliar primaria» reemplazó a «cirrosis biliar primaria» debido a que, en muchos casos, los pacientes no desarrollan cirrosis en las etapas tempranas de la enfermedad, lo que ha llevado a una reevaluación del término original. En lugar de centrarse exclusivamente en la cirrosis, el término actual refleja de manera más precisa los mecanismos fisiopatológicos y la progresión de la enfermedad, que incluyen una inflamación crónica de los conductos biliares intrahepáticos y, eventualmente, la fibrosis hepática.
Desde una perspectiva genética, se ha observado una mayor prevalencia de la enfermedad en los familiares de primer grado de los pacientes afectados, con tasas que oscilan entre el 1,3 % y el 6 %. Este hallazgo sugiere una predisposición genética para desarrollar la enfermedad, lo que se ve reforzado por la identificación de antígenos específicos del sistema HLA, en particular los alelos DRB1*08 y DQB1, que están asociados con una mayor susceptibilidad a la colangitis biliar primaria. Además, el riesgo parece incrementarse en familiares de segundo y tercer grado, y la concordancia en gemelos idénticos es significativamente alta, lo que apunta a una interacción entre factores genéticos y ambientales en la génesis de la enfermedad.
El origen autoinmune de la enfermedad es una de las características más destacadas, ya que la destrucción de los conductos biliares está mediada por una respuesta inmune inapropiada. En este contexto, la colangitis biliar primaria se asocia frecuentemente con otras enfermedades autoinmunes, como el síndrome de Sjögren, las enfermedades tiroideas autoinmunes (como el hipotiroidismo), el síndrome de Raynaud, la esclerosis sistémica (esclerodermia) y la enfermedad celíaca. Esta asociación sugiere un trastorno sistémico en el que el sistema inmunológico del paciente ataca múltiples órganos y tejidos, lo que refuerza la hipótesis de un origen autoinmune compartido.
A pesar de que la causa exacta de la colangitis biliar primaria sigue siendo desconocida, se ha sugerido que ciertos factores infecciosos podrían desempeñar un papel desencadenante en la aparición de la enfermedad. Se ha identificado que infecciones por microorganismos como Novosphingobium aromaticivorans y Chlamydophila pneumoniae pueden inducir o contribuir al desarrollo de la colangitis biliar primaria en individuos susceptibles. Estos agentes infecciosos pueden desencadenar una respuesta inmune que da lugar a la inflamación y destrucción de los conductos biliares.
Además, diversos factores de riesgo ambientales han sido implicados en el desarrollo de la enfermedad. El historial de infecciones urinarias recurrentes, especialmente aquellas causadas por Escherichia coli o Lactobacillus delbrueckii, el tabaquismo, y posiblemente el uso de terapia de reemplazo hormonal y productos como tintes para el cabello han sido asociados con un mayor riesgo de desarrollar la enfermedad. La agrupación temporal y geográfica de casos también sugiere que ciertos factores ambientales pueden tener un papel causal, actuando como disparadores de una predisposición genética o como factores adicionales que favorecen el desarrollo de la enfermedad.
Manifestaciones clínicas
La colangitis biliar primaria es una enfermedad hepática crónica que, en muchos casos, se desarrolla de forma insidiosa, lo que significa que los pacientes pueden ser asintomáticos durante años. Este inicio gradual puede hacer que la enfermedad pase desapercibida durante un largo período, lo que complica el diagnóstico temprano y retrasa el inicio de la intervención terapéutica. Los síntomas iniciales suelen ser sutiles y no específicos, lo que contribuye a esta fase asintomática prolongada. En la mayoría de los pacientes, la manifestación clínica comienza de manera insidiosa, con fatiga como uno de los primeros signos. Esta fatiga a menudo se describe como una somnolencia diurna excesiva, un agotamiento generalizado que no se alivia con el descanso, y que puede afectar de manera significativa la calidad de vida del paciente.
El prurito, o picazón intensa en la piel, es otro síntoma temprano y frecuente, que puede aparecer incluso antes de que se evidencien alteraciones en las pruebas de función hepática. Este síntoma está relacionado con la acumulación de sales biliares en la circulación, lo que provoca una irritación de los nervios cutáneos y una sensación de picor, particularmente en las palmas de las manos y las plantas de los pies. A medida que la enfermedad progresa, se pueden presentar signos más evidentes de alteración hepática.
Con el avance de la enfermedad, y a medida que la destrucción de los conductos biliares intrahepáticos progresa, se puede observar una hepatomegalia (aumento del tamaño del hígado) y esplenomegalia (aumento del tamaño del bazo) durante el examen físico. La hepatomegalia refleja la respuesta inflamatoria y la acumulación de sustancias dentro del hígado, mientras que la esplenomegalia puede ser un indicio de la congestión venosa provocada por la hipertensión portal, un fenómeno que ocurre a medida que la fibrosis hepática progresa. Asimismo, pueden aparecer lesiones xantomatosas, que son depósitos de colesterol que se manifiestan en la piel y los tendones. Estas lesiones son típicamente visibles alrededor de los párpados (conocidas como xantelasmas) y en los tendones, especialmente en los tendones de las manos y los codos. Estas formaciones son un reflejo de la alteración en el metabolismo lipídico asociado a la colestasis.
A medida que la enfermedad avanza hacia etapas más graves, los signos clínicos se vuelven más pronunciados. La ictericia, que es la coloración amarillenta de la piel y las mucosas debido a la acumulación de bilirrubina en sangre, puede aparecer. La esteatorrea, o la presencia de heces grasosas y malolientes debido a la mala absorción de grasas, también puede ser un signo de que el flujo biliar está comprometido. Estos hallazgos clínicos son típicamente indicadores de que la enfermedad ha avanzado a una etapa en la que el daño hepático es más extenso.
En etapas más avanzadas de la enfermedad, pueden aparecer signos de hipertensión portal, como el desarrollo de varices esofágicas, que son dilataciones de los vasos sanguíneos en el esófago causadas por el aumento de la presión en el sistema portal. Aunque las varices esofágicas suelen ser un hallazgo tardío en la colangitis biliar primaria, algunos pacientes pueden presentar estas dilataciones vasculares incluso en fases histológicas tempranas de la enfermedad, lo que indica que la hipertensión portal puede desarrollarse de manera progresiva y, en algunos casos, de forma inesperada.
Además de los síntomas hepáticos y biliares, algunos pacientes con colangitis biliar primaria experimentan alteraciones en el sistema autonómico, que regula funciones involuntarias como la frecuencia cardíaca, la presión arterial y la digestión. La hipotensión ortostática, que es una caída de la presión arterial al ponerse de pie, puede causar mareos y desmayos en los pacientes, mientras que la fatiga asociada puede empeorar la calidad de vida. Además, muchos pacientes presentan síntomas de disfunción cognitiva, como dificultad para concentrarse o problemas de memoria, lo que sugiere que la colangitis biliar primaria podría tener un impacto más amplio en la función del sistema nervioso, más allá de los efectos hepáticos directos.
Un aspecto importante de la enfermedad es su relación con la salud ósea. Los pacientes con colangitis biliar primaria tienen un riesgo aumentado de baja densidad ósea, lo que puede llevar al desarrollo de osteoporosis y un mayor riesgo de fracturas. Este riesgo parece estar asociado con varios factores, incluida la malabsorción de nutrientes esenciales como el calcio y la vitamina D debido a la colestasis crónica, y la posible influencia de polimorfismos en el receptor de vitamina D, lo que puede alterar la capacidad del cuerpo para metabolizar y utilizar la vitamina D de manera eficaz. La deficiencia de vitamina D es un factor clave en la salud ósea, y la alteración en su metabolismo en pacientes con colangitis biliar primaria puede contribuir a una mayor fragilidad ósea y a un mayor riesgo de fracturas.
Exámenes diagnósticos
En las primeras etapas de la colangitis biliar primaria, los análisis de sangre pueden ser completamente normales, lo que dificulta el diagnóstico precoz de la enfermedad. Esto se debe a que, en sus fases iniciales, la enfermedad puede no producir alteraciones significativas en las pruebas de función hepática convencionales, lo que permite que el paciente permanezca asintomático o con síntomas mínimos. A medida que la enfermedad progresa y la destrucción de los conductos biliares intrahepáticos se intensifica, se observa una alteración en las pruebas bioquímicas hepáticas, las cuales reflejan una colestasis, es decir, una disminución del flujo biliar.
En las fases más avanzadas de la enfermedad, se detectan niveles elevados de fosfatasa alcalina, un marcador clave de obstrucción biliar intrahepática. Este aumento en la fosfatasa alcalina es uno de los primeros indicios de que está ocurriendo colestasis, incluso antes de que otros signos clínicos o pruebas de función hepática sean patológicos. Además, el colesterol, especialmente el colesterol lipoproteína de alta densidad (HDL) y la lipoproteína X, también se encuentra elevado. La lipoproteína X es una lipoproteína anómala que aparece en la colestasis crónica y se asocia con la acumulación de bilis en la circulación, lo que contribuye a la alteración en los perfiles lipídicos. A medida que la enfermedad avanza, se observa un aumento en los niveles de bilirrubina, el pigmento biliar que se acumula en la sangre cuando el flujo biliar está comprometido. Esto puede llevar a la aparición de ictericia, la cual es un signo clínico característico de la progresión hacia etapas más graves de la enfermedad.
Un hallazgo serológico clave en el diagnóstico de la colangitis biliar primaria es la presencia de anticuerpos antimitocondriales en un porcentaje muy alto de los pacientes, aproximadamente en el 95% de los casos. Estos anticuerpos son específicos para la enfermedad y sirven como un marcador serológico esencial en el diagnóstico. Los anticuerpos antimitocondriales son dirigidos contra componentes de los mitocondrios, específicamente contra la proteína PDC-E2 (dihidrolipoil-transacetilasa del complejo de la piruvato deshidrogenasa), que está involucrada en el metabolismo de los ácidos grasos y la producción de energía en las células hepáticas. La presencia de estos anticuerpos es casi patognomónica de la colangitis biliar primaria, y su hallazgo en suero con un título superior a 1:40 es uno de los criterios diagnósticos clave. Además, los niveles de inmunoglobulina M (IgM) suelen estar elevados en los pacientes con colangitis biliar primaria, lo que también puede ser útil para el diagnóstico. Estos aumentos en las concentraciones de IgM son una manifestación de la respuesta inmune anómala que caracteriza a la enfermedad.
El diagnóstico de colangitis biliar primaria se establece principalmente mediante la combinación de pruebas serológicas y bioquímicas. Las químicas hepáticas colestáticas, como la elevación de la fosfatasa alcalina y los anticuerpos antimitocondriales detectados en el suero, son fundamentales para la identificación de la enfermedad. No obstante, en algunos casos, es recomendable realizar una ecografía de base para evaluar la anatomía del hígado y la presencia de signos indirectos de enfermedad hepática, como el aumento de tamaño del órgano o la alteración de la estructura de los conductos biliares.
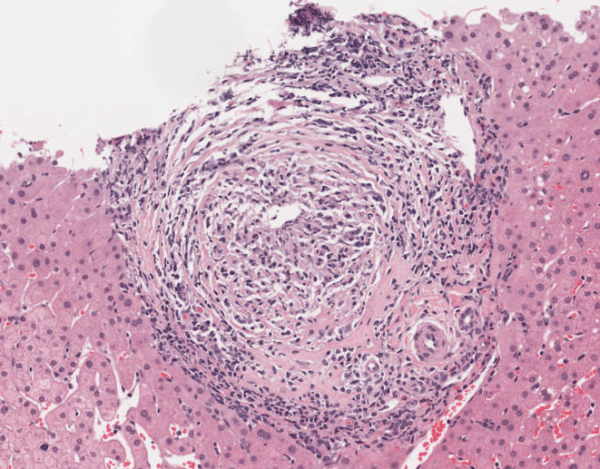
Aunque la biopsia hepática no es estrictamente necesaria para el diagnóstico de la colangitis biliar primaria, salvo en los casos en los que los anticuerpos antimitocondriales estén ausentes, sigue siendo una herramienta útil para evaluar la extensión del daño hepático y la estadificación histológica de la enfermedad. La biopsia hepática permite clasificar la enfermedad en diferentes estadios, según los hallazgos histológicos observados. El estadio I se caracteriza por la presencia de inflamación portal acompañada de granulomas, estructuras inflamatorias típicas de la enfermedad. El estadio II muestra una proliferación de conductos biliares y inflamación periportal, es decir, alrededor de los conductos biliares. En el estadio III, se observan septos fibrosos interlobulillares, que son signos de fibrosis avanzada en el hígado. Finalmente, el estadio IV corresponde a la presencia de cirrosis, que es el daño hepático irreversible debido a la fibrosis extensa y la formación de nódulos hepáticos.
Aunque la biopsia hepática proporciona información valiosa sobre el grado de daño hepático, existen métodos no invasivos que también pueden ayudar a estimar el grado de fibrosis hepática. Uno de estos métodos es el análisis de fibrosis hepática mejorada (ELF, por sus siglas en inglés), que mide los niveles séricos de ciertos biomarcadores, como el ácido hialurónico, el inhibidor de la metaloproteinasa-1 y el procóligo III aminopéptido, que reflejan el grado de daño y remodelación del tejido hepático. Además, técnicas de elastografía por ultrasonido o resonancia magnética(RM) son cada vez más utilizadas para evaluar la rigidez hepática, lo que es un indicio indirecto de fibrosis y cirrosis. Estas tecnologías proporcionan información detallada sobre la rigidez del hígado, lo que permite una evaluación precisa de la progresión de la fibrosis sin necesidad de realizar una biopsia.
Diagnóstico Diferencial
La colangitis biliar primaria es una enfermedad hepática autoinmune crónica, pero su diagnóstico debe realizarse cuidadosamente para diferenciarla de otras afecciones que pueden presentar un cuadro clínico e incluso hallazgos histológicos similares. Esta diferencia es crucial para asegurar el tratamiento adecuado, ya que las opciones terapéuticas varían dependiendo de la causa subyacente. Las condiciones que deben ser diferenciadas de la colangitis biliar primaria incluyen, entre otras, la obstrucción crónica de las vías biliares, el carcinoma de los conductos biliares, la colangitis esclerosante primaria, la sarcoidosis, la toxicidad colestática inducida por fármacos y, en algunos casos, la hepatitis crónica.
Uno de los diagnósticos diferenciales más importantes es la obstrucción crónica de las vías biliares, que puede ser causada por litos (cálculos biliares) o estenosis (estrechamiento de los conductos biliares). En estos casos, la obstrucción del flujo biliar genera una colestasis que puede producir síntomas y alteraciones bioquímicas similares a los de la colangitis biliar primaria, como la elevación de la fosfatasa alcalina y la ictericia. Sin embargo, la causa de la obstrucción es mecánica y no autoinmune, lo que hace que el tratamiento deba centrarse en la resolución de la obstrucción, como la extracción de los cálculos o la dilatación de los conductos, en lugar de la inmunosupresión que se emplea en la colangitis biliar primaria.
El carcinoma de los conductos biliares, también conocido como colangiocarcinoma, es otro diagnóstico diferencial importante, ya que puede presentarse con colestasis crónica y síntomas similares, como fatiga, ictericia y pérdida de peso. A diferencia de la colangitis biliar primaria, el carcinoma de los conductos biliares es un tumor maligno que requiere un enfoque terapéutico completamente diferente, que incluye cirugía, quimioterapia o radioterapia, dependiendo de su estadio.
La colangitis esclerosante primaria es una enfermedad autoinmune crónica que también involucra la inflamación y fibrosis de los conductos biliares, pero a diferencia de la colangitis biliar primaria, esta enfermedad suele estar asociada con una enfermedad inflamatoria intestinal, especialmente la colitis ulcerosa. La colangitis esclerosante primaria afecta principalmente a los conductos biliares grandes, a menudo con una característica patrón de estenosis y dilatación alternadas de los conductos, que se puede visualizar mediante pruebas de imagen, como la colangiopancreatografía retrógrada endoscópica o la colangiografía por resonancia magnética. Por otro lado, la colangitis biliar primaria se caracteriza por la destrucción de los conductos biliares intrahepáticos pequeños sin que se observe la misma afectación de los conductos biliares grandes.
La sarcoidosis es una enfermedad inflamatoria multisistémica que se caracteriza por la formación de granulomas en varios órganos, incluidos el hígado y los conductos biliares. Aunque la sarcoidosis puede causar colestasis y anomalías en las pruebas de función hepática, la presencia de granulomas en el hígado es un hallazgo distintivo que la diferencia de la colangitis biliar primaria. Además, en la sarcoidosis, los granulomas son generalmente no caseosos, lo que los distingue de otras causas inflamatorias hepáticas.
La toxicidad colestática inducida por fármacos también es una causa importante a considerar, ya que varios medicamentos, como la clorpromazina y otros fármacos antipsicóticos, pueden inducir una colestasis similar a la que se observa en la colangitis biliar primaria. Sin embargo, en estos casos, la suspensión del medicamento responsable generalmente conduce a una mejoría de los síntomas y de las pruebas bioquímicas, lo que puede ayudar a diferenciar esta causa de la colangitis biliar primaria, que es una enfermedad autoinmune crónica que no mejora con la eliminación de un agente externo.
La hepatitis crónica, tanto de causa viral como autoinmune, también debe diferenciarse de la colangitis biliar primaria, ya que puede presentar alteraciones en las pruebas de función hepática y síntomas como fatiga e ictericia. Sin embargo, la hepatitis crónica típicamente involucra un daño más difuso al parénquima hepático y no está asociada con la destrucción específica de los conductos biliares intrahepáticos, lo que permite una distinción con la colangitis biliar primaria.
En algunos casos, los pacientes pueden presentar un cuadro clínico e histológico compatible con la colangitis biliar primaria pero sin la presencia de los anticuerpos antimitocondriales, que son uno de los principales marcadores diagnósticos de la enfermedad. A estos pacientes se les denomina colangitis biliar primaria negativa para anticuerpos antimitocondriales. Este subtipo, que previamente se conocía como colangitis autoinmune, está asociado con niveles más bajos de inmunoglobulina M en suero y una mayor frecuencia de otros anticuerpos autoinmunes, como los anticuerpos contra músculo liso y anticuerpos antinucleares (ANA). En estos pacientes, aunque los anticuerpos antimitocondriales no se detectan mediante las pruebas convencionales, a menudo pueden encontrarse mediante técnicas más avanzadas, como la inmunoblot contra proteínas recombinantes. Esta técnica más sensible puede identificar anticuerpos antimitocondriales que no son detectados por las pruebas de inmunofluorescencia estándar.
Finalmente, algunos pacientes pueden presentar características superpuestas de colangitis biliar primaria y hepatitis autoinmune, una condición conocida como hepatitis autoinmune-colangitis biliar primaria superpuesta. En estos casos, los pacientes tienen un cuadro clínico e histológico que cumple los criterios para ambas enfermedades, lo que puede complicar el diagnóstico y el manejo. Esta superposición sugiere que tanto la colangitis biliar primaria como la hepatitis autoinmune comparten mecanismos patogénicos comunes, como la activación inapropiada del sistema inmunológico, y puede requerir un enfoque terapéutico más complejo que aborde ambas afecciones.
Tratamiento
El tratamiento del prurito en los pacientes con colangitis biliar primaria (CBP) representa un desafío significativo debido a la naturaleza crónica de la enfermedad y la resistencia que algunos pacientes pueden mostrar a los tratamientos convencionales. El prurito, uno de los síntomas más molestos asociados a la colestasis, se origina por la acumulación de ácidos biliares en la piel debido a la obstrucción del flujo biliar, lo que lleva a la estimulación de los receptores pruríticos. Para manejar este síntoma, existen varias opciones terapéuticas, algunas de las cuales pueden ser más efectivas que otras, dependiendo del paciente y de la severidad del prurito.
La colestiramina es uno de los tratamientos más comunes para el prurito asociado con la colestasis. Este medicamento es una resina intercambiadora de aniones que se administra por vía oral, generalmente en dosis de 4 gramos, tres veces al día, disuelta en agua o jugo. Su mecanismo de acción se basa en la unión de los ácidos biliares en el intestino, evitando que sean reabsorbidos en el torrente sanguíneo y, de esta manera, reduciendo su concentración en el cuerpo. Esta reducción de ácidos biliares circulantes disminuye la estimulación de los receptores pruríticos en la piel, aliviando el prurito. Sin embargo, es importante tener en cuenta que la colestiramina puede interferir con la absorción de vitaminas liposolubles (A, D y K), lo que puede llevar a deficiencias vitamínicas, especialmente si el paciente presenta esteatorrea, una condición que dificulta la absorción de grasas y vitaminas liposolubles debido a la colestasis.
Existen otros secuestradores de ácidos biliares, como el colestipol y el colesevelam, que son similares a la colestiramina en su función, pero tienden a ser mejor tolerados por algunos pacientes debido a su menor incidencia de efectos secundarios gastrointestinales. No obstante, a pesar de su mejor tolerancia, no se ha demostrado que estos fármacos reduzcan el prurito de manera tan efectiva como la colestiramina.
Por otro lado, la rifampicina, un antibiótico utilizado principalmente para tratar la tuberculosis, ha demostrado tener cierto efecto en el alivio del prurito en algunos pacientes con CBP, aunque los resultados son inconsistentes. La rifampicina actúa probablemente al inducir la excreción de ácidos biliares a través del aumento de la actividad de la glucuroniltransferasa, enzima que facilita la conjugación de los ácidos biliares. Sin embargo, debido a la variabilidad en su eficacia, no se considera un tratamiento de primera línea.
En cuanto a los antagonistas opioides, como naloxona y naltrexona, hay creciente evidencia de que pueden ser útiles en el manejo del prurito, dado que los opioides endógenos pueden jugar un papel importante en la mediación del prurito. Estos fármacos antagonizan los efectos de los opioides sobre los receptores del sistema nervioso central, lo que puede reducir la sensación de prurito en pacientes con colestasis. Sin embargo, uno de los riesgos asociados con el uso de estos antagonistas opioides es que pueden inducir síntomas de abstinencia de opioides, especialmente si se administran en pacientes que ya tienen una exposición a opioides o en aquellos que son particularmente sensibles a los efectos de estos medicamentos. La dosificación de naloxona puede ser 0.2 microgramos por kilogramo por minuto por vía intravenosa, o naltrexona puede iniciarse a una dosis de 12.5 miligramos por día por vía oral.
Otros medicamentos que han mostrado eficacia en el tratamiento del prurito en pacientes con colangitis biliar primaria incluyen ondansetrón, un antagonista del receptor de serotonina 5-hidroxitriptamina (5-HT3), y sertralina, un inhibidor selectivo de la recaptación de serotonina (ISRS). Ondansetrón, administrado a dosis de 4 miligramos por vía oral tres veces al día según sea necesario, y sertralina, con una dosis de 75 a 100 miligramos por día, pueden ser útiles en algunos pacientes, dado que se ha demostrado que los niveles elevados de serotonina en el sistema nervioso central pueden estar relacionados con el prurito. Además, gabapentina, un medicamento anticonvulsivo, ha mostrado cierto beneficio en algunos pacientes, administrándose en dosis de 300 a 2400 miligramos diarios.
Para los casos de prurito refractario, es decir, aquellos que no responden a los tratamientos farmacológicos convencionales, se pueden considerar procedimientos más invasivos, como la plasmapheresis o la diálisis extracorpórea de albúmina. Estos procedimientos permiten la eliminación de sustancias pruriginosas del plasma sanguíneo, como los ácidos biliares, lo que puede aliviar los síntomas en algunos pacientes, aunque no está exento de riesgos.
En cuanto a la somnolencia diurna, un síntoma común en pacientes con colangitis biliar primaria, se ha encontrado que modafinil, un medicamento que promueve la vigilia, puede ser útil. La dosificación habitual de modafinil es de 100 a 200 miligramos diarios por vía oral. Sin embargo, este fármaco puede no ser bien tolerado en todos los pacientes, lo que limita su uso en algunos casos.
El tratamiento farmacológico de la colangitis biliar primaria está dirigido a reducir la colestasis y, en consecuencia, mejorar la función hepática y la calidad de vida de los pacientes. El ácido ursodeoxicólico es el tratamiento de elección para esta enfermedad y se utiliza en dosis de 13 a 15 miligramos por kilogramo de peso corporal al día, generalmente dividido en dos dosis. El ácido ursodeoxicólico ha demostrado ser eficaz para reducir la progresión de la enfermedad, especialmente en las etapas tempranas. En estudios clínicos, se ha observado que el ácido ursodeoxicólico estabiliza la histología hepática, mejora la supervivencia a largo plazo, reduce el riesgo de desarrollar varices esofágicas y retrasa o incluso previene la necesidad de un trasplante hepático. Aunque no siempre se logra una normalización completa de las pruebas bioquímicas hepáticas, el tratamiento con ácido ursodeoxicólico ha mostrado beneficios a largo plazo en la supervivencia de los pacientes, particularmente en aquellos con enfermedad en estadio 1 o 2. En algunos pacientes que no responden adecuadamente a este tratamiento o que lo toleran mal, se puede considerar el uso de ácido obeticólico, un agonista del receptor X de farnesoides, que puede añadirse al tratamiento en caso de respuesta incompleta o intolerancia al ácido ursodeoxicólico.
Pronóstico
La colangitis biliar primaria es una enfermedad hepática crónica y progresiva que, sin intervención médica adecuada, puede llevar a insuficiencia hepática terminal y la necesidad de un trasplante de hígado. En ausencia de trasplante hepático, la supervivencia promedio de los pacientes con colangitis biliar primaria es de aproximadamente 7 a 10 años una vez que se desarrollan los síntomas clínicos de la enfermedad. Este pronóstico está influenciado por varios factores, incluyendo la etapa de la enfermedad en el momento del diagnóstico, el tratamiento recibido, y las comorbilidades asociadas.
El tratamiento con ácido ursodeoxicólico, que se ha introducido como terapia estándar para la colangitis biliar primaria, ha tenido un impacto significativo en la supervivencia, especialmente en pacientes diagnosticados en etapas tempranas de la enfermedad. Se ha observado que las mujeres jóvenes que inician el tratamiento con ácido ursodeoxicólico tienen una supervivencia mejorada en comparación con las que no reciben dicho tratamiento. Este medicamento ayuda a reducir la colestasis y a mejorar la función hepática, lo que ralentiza la progresión hacia la insuficiencia hepática y, en algunos casos, estabiliza la enfermedad, lo que contribuye a una expectativa de vida más larga. La mejora es particularmente notable en los casos de diagnóstico temprano, antes de que se desarrollen complicaciones graves como la cirrosis.
La progresión hacia insuficiencia hepática y hipertensión portal (una complicación de la cirrosis) puede acelerarse si existen factores adicionales que favorezcan la evolución negativa de la enfermedad. El tabaquismo, por ejemplo, se ha identificado como un factor de riesgo significativo que puede empeorar los resultados de los pacientes con colangitis biliar primaria. Fumar puede aumentar la inflamación hepática y contribuir a la fibrosis, lo que acelera la progresión hacia la cirrosis. Además, los estudios han mostrado que los hombres con colangitis biliar primaria tienden a tener peores resultados que las mujeres, lo que sugiere que el sexo juega un papel en la evolución de la enfermedad, aunque las razones exactas para esta diferencia aún no están completamente claras. Posiblemente, las diferencias hormonales y la respuesta inmune podrían ser factores subyacentes.
En términos de pronóstico, los pacientes con colangitis biliar primaria que se encuentran en etapas tempranas de la enfermedad tienen una mejor esperanza de vida si responden adecuadamente al tratamiento con ácido ursodeoxicólico. Un indicador importante de buen pronóstico es el nivel de fosfatasa alcalina y aspartato aminotransferasa (AST), que deben ser menores de 1.5 veces el valor normal, y la bilirrubina total debe ser de 1 mg/dL (17.1 micromoles por litro) o menos después de un año de tratamiento. Estos criterios, conocidos como criterios de París II, han demostrado correlacionarse con un bajo riesgo de progresión hacia la cirrosis y una supervivencia similar a la de la población sana a largo plazo. Los pacientes que cumplen con estos criterios tienen una probabilidad significativamente menor de desarrollar complicaciones graves relacionadas con la enfermedad hepática, como la hipertensión portal o la necesidad de un trasplante hepático.
Sin embargo, cuando la enfermedad progresa a etapas más avanzadas y los pacientes desarrollan insuficiencia hepáticasevera, el trasplante hepático se convierte en la única opción terapéutica viable. En estos casos, la decisión de realizar un trasplante debe basarse en una evaluación exhaustiva de la gravedad de la enfermedad, lo que generalmente se mide a través de diversas puntuaciones de evaluación. El MELD-Na (Model for End-Stage Liver Disease con la variable de sodio) es una de las puntuaciones más utilizadas para determinar la urgencia de un trasplante. Si la puntuación MELD-Na alcanza 15 o más, o si la bilirrubina total en suero es de 6 mg/dL (102.6 micromoles por litro) o más, o si la puntuación Mayo (que evalúa la probabilidad de mortalidad a corto plazo en pacientes con colangitis biliar primaria) es 7.8 o mayor, el trasplante hepático debe considerarse como una opción terapéutica urgente. Estos criterios indican un pronóstico de mortalidad a corto plazo elevado, lo que hace necesario intervenir con un trasplante hepático para salvar la vida del paciente.

Fuente y lecturas recomendadas:
Abbas N et al. UK-wide multicenter evaluation of second-line therapies in primary biliary cholangitis. Clin Gastroenterol Hepatol. 2023;21:1561. [PMID: 35961518]
Bhan I et al. Case 4-2023: a 56-year-old man with abnormal results on liver testing. N Engl J Med. 2023;388:544. [PMID: 36780679]
Khakoo NS et al. Efficacy and safety of bezafibrate alone or in combination with ursodeoxycholic acid in primary biliary cholangitis: systematic review and meta-analysis. Dig Dis Sci. 2023;68:1559. [PMID: 36180756]
Kowdley KV et al. Application of the latest advances in evidencebased medicine in primary biliary cholangitis. Am J Gastroenterol. 2023;118:232. [PMID: 36729104]
Trivella J et al. Primary biliary cholangitis: epidemiology, prognosis, and treatment. Hepatol Commun. 2023;7:e0179. [PMID: 37267215]
Aprende administración paso a paso

ADMINISTRACION DESDE CERO
Originally posted on 16 de enero de 2025 @ 9:06 AM